
Миотоническая мышечная дистрофия болезнь штейнерта

Дегенеративно-дистрофические заболевания нервной системы с преобладающим поражением периферических нервов и мышечных волокон занимают огромную долю в структуре наследственной патологии человека.
Типичным представителем является миотоническая дистрофия (или дистрофическая миотония), описанная в начале прошлого века несколькими авторами и получившая название болезни Россолимо-Штейнерта-Куршмана.
Этот недуг является самым известным заболеванием из разряда миотоний и самой распространенной формой мышечной дистрофии у взрослых людей. Что представляет собой эта болезнь и как с ней бороться?
Открытие и суть заболевания
Россолимо, Штейнерт и Куршман изучали болезнь, являющуюся генетической патологией с аутосомно-доминантным типом наследования. Это значит, что один родитель имеет мутантный ген, больные дети при этом рождаются с вероятностью 50%. Заболевание носит характер семейного недуга и передается последующим поколениям по вертикали.
Сыновья и дочери в таких семьях болеют с одинаковой частотой, примерно 3 — 5 человек на 100 тысяч населения. Возраст начала заболевания, а также выраженность симптомов отличаются значительной вариабельностью.
Описаны ранние неонатальные и поздние формы, однако чаще всего заболевание дебютирует на втором, реже — на третьем десятке жизни. 
Отмечено, что передача болезни ребенку от матери является более прогностически неблагоприятной, чем от отца.
В основе болезни лежит дефект гена из 19 пары хромосом, который отвечает за синтез фермента миотонин-протеинкиназы. Это белок в норме присутствует не только в скелетной мускулатуре, но и в клетках миокарда и ЦНС.
Вот почему для дистрофической миотонии характерна полисистемность проявлений с поражением разных органов и систем. Неполноценность миотонин-протеинкиназы приводит к появлению мышечных спазмов вместе с атрофическими изменениями мускулатуры головы, шейного отдела, конечностей.
Наблюдается сочетание гипертрофии одних мышечных волокон с атрофией других и заменой их на жировую или соединительную ткани.
Клинические проявления
В связи с варьированием начала заболевания в клинике различают следующие формы по возрастному принципу:
- врожденная форма — манифестация болезни начинается сразу после появления ребенка на свет;
- юношеский вариант — дебют миотонии в возрасте от одного года до периода полового созревания;
- классическая форма — начало клинических проявлений приходится на второй и третий десяток жизни;
- минимальный вариант — манифестация приходится на поздние сроки — шестой десяток жизни.
Характерно, что чем позднее проявляется болезнь, тем благоприятнее течение и лучше прогноз. Чаще всего встречается классическая форма болезни Штейнерта, для которой типичными являются следующие клинические симптомы:
- Миотония — проявляется спазмами жевательных мышц и сгибателей кистей рук, характерны атрофические изменения в разных
![]()
группах мышц. Постепенно происходит угасание миотонических проявлений и прогрессирования мышечной дистрофии, внешне это выражается в печальной маске лица и отсутствии мимики. Опасным является парез мышц гортани с нарушением глотания, а также слабость дыхательной мускулатуры, в результате возможны приступы остановки дыхания во сне, развитие пневмонии. - Сердечнососудистые нарушения — нарушения ритма сердца, гипертрофические изменения левого желудочка, выявляемые на ЭКГ, застойная сердечная недостаточность.
- Эндокринные расстройства (в основном затрагиваются половые функции) — уменьшение размеров половых органов, снижение сексуального влечения, у женщин — расстройства менструального цикла, ожирение.
- Общие изменения дистрофического характера — сухость и пигментация кожных покровов, выпадение частично или полностью волос и зубов, ранняя катаракта.
- Нарушения со стороны ЦНС — усталость, расстройства сна, апатия, потеря интеллекта.

Отдельно стоит отметить характерные клинические проявления врожденной формы дистрофической миопатии:
- уменьшение активных движений плода в утробе матери, выявляемое во время УЗИ;
- в период новорожденности — вялость, распространенная гипотония, особенно в жевательных, мимических, мышцах глазных яблок;
- сохранение и даже повышение сухожильных рефлексов;
- проблемы вскармливания, расстройства дыхания по типу респираторного дистресс-синдрома;
- задержка физического и нервно-психического развития, признаки олигофрении;
- стремительное прогрессирование заболевания, высокий риск внезапной смерти.
Диагностические критерии
Подозрение на болезнь Россолимо-Штейнерта — Куршмана может возникнуть у врача при наличии у пациента сочетания миотонических и дистрофических изменений в мышцах на фоне потери интеллекта и наличия сердечнососудистой и эндокринной патологии.
Полисистемность практически всегда свидетельствует о генетической природе заболевания. Такие больные подлежат анализу ДНК и проведению генеалогического анализа для подтверждения аутосомно-доминантного наследования патологии. В качестве информативных методов исследования используются электрокардиография, электронейромиография, анализы на гормоны.
В связи с многогранностью клинических проявлений к процессу постановки диагноза обычно привлекаются специалисты из разных отраслей медицины — генетики, кардиологии, эндокринологии, гинекологии, андрологии, неврологии.
Дифференциальный диагноз проводится между дистрофической миотонией и другими видами схожих заболеваний. В отличие от остальных для болезни Россолимо характерна мышечная атрофия. Нередко для подтверждения диагноза приходится прибегать к биопсии, чтобы определить уровень мышечного белка, который в тканях при данной патологии повышен.
Проводится также антенатальная диагностика методом исследования околоплодных вод.

Медицинская помощь
Генетическое заболевание невозможно излечить полностью, поэтому целью лечения при болезни Россолимо-Штейнерта-Куршмана является купирование симптомов, улучшение общего состояния и социальная адаптация пациентов.
Принципы лечения заключаются в следующем:
- диета с низким содержанием солей калия (яблоки, спаржа, капуста, огурцы, виноград, зелень, кукуруза, ягоды, редис, мандарины,
![]()
грейпфрут, лук, морковь, баклажаны, горох); - исключение переохлаждений во избежание возникновения спазмов;
- применение препаратов хинина для стабилизации клеточных мембран, таких лекарств, как Дифенин, Новокаинамид, Диакарб — для снятия мышечных спазмов и уменьшения скованности, судорог, снижения внутричерепного давления;
- использование анаболических стероидов (Метанандростенолон, Ретаболил, Нерабол), витаминов группы В, АТФ для стимулирования мышечной массы;
- ЛФК, массаж, электромиостимуляция, ортопедические приспособления.

Перечисленные мероприятия дают хороший положительный эффект как при классической, так и при врожденной форме болезни. Полностью избавить больного от дистрофической миотонии они не могут, но продлить ему жизнь и улучшить ее качество способны.
Прогноз хуже у врожденной формы — летальность высока, дети могут не дожить до 3 лет. Юношеский вариант миотонии протекает достаточно 
тяжело и может привести уже в молодые годы к ограничению трудоспособности и ранней инвалидности.
В случае классической формы заболевание может протекать долго при проведении своевременных лечебно-коррекционных мероприятий. Наиболее благоприятный прогноз у поздно проявившихся форм болезни.
Профилактические мероприятия сводятся к тому, что женщине из семьи с неблагополучным анамнезом на стадии планирования беременности необходимо пройти обследование на наличие аномальных генов, ответственных за развитие мышечной дистрофии. Это целесообразно сделать также в случае наличия у родственников отца ребенка данной патологии.
Возможности к рождению деток должны решаться индивидуально в каждом конкретном случае врачами — генетиками после консилиума.

Дистрофическая миотония Россолимо-Штейнерта-Куршмана — наследственное медленно прогрессирующее заболевание, в основе которого лежит дефект миотонин-протеинкиназы, приводящий к развитию миотонии в сочетании с дистрофическими изменениями мышечной ткани. Заболевание проявляется миотоническими спазмами, атрофическими изменениями мышц шеи, лица и дистальных отделов конечностей, снижением интеллекта, аритмиями и эндокринной патологией. Диагностика дистрофической миотонии основывается на клинических данных, результатах генеалогического анализа и исследования ДНК. Лечение симптоматическое, направленное против симптомов миотонии (фенитоин, прокаинамид, хинин, мочегонные) и мышечной дистрофии (анаболические стероиды, АТФ).
МКБ-10
- Причины
- Симптомы классической формы
- Симптомы врожденной формы
- Диагностика
- Лечение миотонии Россолимо-Штейнерта-Куршмана
- Цены на лечение
Общие сведения
Дистрофическая миотония Россолимо-Штейнерта-Куршмана является наследственным заболеванием и передается от родителей к детям по аутосомно-доминантному типу. Классическая форма этого заболевания развивается преимущественно в возрастном периоде от 10 до 20 лет. В более редких случаях встречается врожденная дистрофическая миотония Россолимо-Штейнерта-Куршмана, клинические симптомы которой проявляются сразу же после рождения.
Морфологически при миотонии Россолимо-Штейнерта-Куршмана отмечается сочетание гипертрофических изменений одних мышечных волокон с атрофией других, замещение части мышечных волокон жировой и соединительной тканью. Изучение образцов мышечной ткани под электронным микроскопом показывает деструкцию миофибрилл и изменение размера митохондрий.
Причины
Последние исследования генетического набора больных дистрофической миотонией показали, что основу заболевания составляет дефект в гене DMPK, находящемся в 19-й хромосоме и отвечающем за синтез миотонин-протеинкиназы. У больных дистрофической миотонией выявляется значительное увеличение тринуклеотидных CTG-повторов в основной части гена DMPK. При этом именно от количества повторов зависит форма и тяжесть миотонии.
В норме число тринуклеотидных повторов варьирует от 5 до 37. Увеличение повторов до 50-80 приводит к появлению мягкой формы миотонии Россолимо-Штейнерта-Куршмана. Если количество тринуклеотидных повторов находится в промежутке от 100 до 500, развивается поздняя форма заболевания. Врожденные формы дистрофической миотонии возникают при повышении числа CTG-повторов от 500 до 2000. Исследования показали, что увеличение тринуклеотидных повторов происходит в основном в женских гаметах в процессе мейоза. В связи с этим при передаче заболевания от матери у ребенка возникает более тяжелая форма миотонии или ее врожденный вариант.
Симптомы классической формы
В классическом варианте миотония Россолимо-Штейнерта-Куршмана начинает проявляться после первых 5 лет жизни и может манифестировать до 35-летнего возраста. Но наиболее часто клинические проявления заболевания возникают в возрастном диапазоне от 10 до 20 лет. Они представляют собой сочетание типичных симптомов миотонии с признаками миопатии, поражением сердечно-сосудистой системы и ЦНС, эндокринными нарушениями и катарактой.
Из миотонических проявлений для миотонии Россолимо-Штейнерта-Куршмана характерны миотонические спазмы, наиболее выраженные в жевательных мышцах и мышцах-сгибателях кисти. Наблюдаются также механические реакции миотонического типа, выявляемые при ударе неврологическим молоточком. Отличительной особенностью миотонии Россолимо-Штейнерта-Куршмана является наличие атрофических изменений в различных группах мышц. При этом течение заболевания характеризуется постепенным угасанием симптомов миотонии на фоне прогрессирующей мышечной дистрофии.
Чаще всего при миотонии Россолимо-Штейнерта-Куршмана поражаются мышцы дистальных отделов конечностей, мимическая мускулатура, грудино-ключично-сосцевидные и височные мышцы. Поражение мимических мышц проявляется характерным маскообразным печальным выражением лица больных дистрофической миотонией. Атрофические изменения мышц глотки и гортани приводят к развитию миопатического пареза гортани с нарушением голоса и затруднением глотания. Миопатические изменения могут возникать в дыхательной мускулатуре. Наряду с миотоническими спазмами они приводят к ухудшению легочной вентиляции, появлению приступов апноэ во сне, возникновению застойной или аспирационной пневмонии.
Нарушения сердечно-сосудистой системы наблюдаются примерно в половине случаев дистрофической миотонии. К ним относятся аритмии, связанные с нарушением проводимости, и гипертрофия левого желудочка. Наиболее распространена блокада ножек пучка Гиса. Из признаков поражения ЦНС чаще всего наблюдается гиперсомния и снижение интеллектуальных способностей, доходящее до легкой степени дебильности.
Эндокринные расстройства при миотонии Россолимо-Штейнерта-Куршмана затрагивают в основном половую сферу. У мужчин они проявляются снижением либидо, крипторхизмом, импотенцией, гипогонадизмом, у женщин — гирсутизмом, нарушениями менструального цикла (олигоменореей, дисменореей) и ранним климаксом. Типичным является изменение структуры волос в сочетании с алопецией. У мужчин отмечается выпадение волос на висках и в области лба, у женщин — диффузное или очаговое облысение.
Симптомы врожденной формы
Первые признаки врожденной формы миотонии Россолимо-Штейнерта-Куршмана могут проявляться еще в период внутриутробного развития. Как правило, они выражаются в значительном снижении двигательной активности плода, которое диагностируется акушером-гинекологом по данным акушерского УЗИ в III триместре беременности.
После рождения ребенка преобладают симптомы миопатии. Отмечается диффузная гипотония мышц, более выраженная в мимической, жевательной и глазодвигательной мускулатуре, а также в мышечных группах дистальных отделов конечностей. Характерны затруднения вскармливания и дыхательные расстройства. Миотоническая симптоматика начинает проявляться несколько позже. Врожденная дистрофическая миотония сопровождается задержкой моторного развития и олигофренией. Типично быстрое прогрессирование симптомов заболевания, часто приводящее к смертельному исходу еще в раннем детстве.
Диагностика
Типичное сочетание миотонии с признаками дистрофических изменений мышечной ткани, умственной отсталостью, нарушениями со стороны сердечно-сосудистой и эндокринной систем позволяет неврологу предположить миотонию Россолимо-Штейнерта-Куршмана. Подтверждением диагноза являются результаты генеалогического анализа, свидетельствующие об аутосомно-доминантном типе наследования заболевания, и данные ДНК-анализа. Дополнительно проводится электромиография, электронейрография, исследования половых гормонов, ЭКГ. К диагностике пациентов с миотонией Россолимо-Штейнерта-Куршмана могут дополнительно привлекаться генетики, кардиологи, эндокринологи, гинекологи, андрологи.
При диагностике дистрофической миотонии ее необходимо дифференцировать ее от других видов миотонии. Так, наличие мышечных атрофий позволяет отличить миотонию Россолимо-Штейнерта-Куршмана от миотонии Томсена, для которой типична мышечная гипертрофия. От миотонии Беккера заболевание отличается ранним поражением мышц лица и доминантным типом наследования. Кроме того, следует проводить дифференциальный диагноз миотонии Россолимо-Штейнерта-Куршман с миопатиями, БАС и амиотрофией Шарко-Мари-Тута.
Лечение миотонии Россолимо-Штейнерта-Куршмана
Радикальной терапии миотонии Россолимо-Штейнерта-Куршмана пока не существует. Пациентам, имеющим это заболевание, показана диета со сниженным содержанием калия. Им также следует избегать переохлаждения, которое провоцирует миотонические спазмы. Уменьшению миотонических проявлений способствует прием хинина, прокаинамида, фенитоина в сочетании с ацетазоламидом. Показаны анаболические стероиды ( нандролона деканоат, метиландростендиол, метандростенол), небольшие дозы АТФ, витамины группы В.
Миотоническая дистрофия (болезнь Штейнерта) — 2-я по частоте встречаемости мышечная дистрофия в Северной Америке, Европе и Австралии. Заболеваемость составляет 1:30 000 общей популяции. Тип наследования — аутосомно-доминантный.
Миотоническая дистрофия — пример заболевания, при котором генетический дефект вызывает дисфункцию многих органов. Поражаются не только поперечнополосатые мышцы, но также гладкая мускулатура ЖКТ и матки, нарушается функция сердца, выявляются множественные и вариабельные эндокринопатии, иммунодефицит, катаракта, дизморфия лица, снижение интеллекта и другие неврологические нарушения.
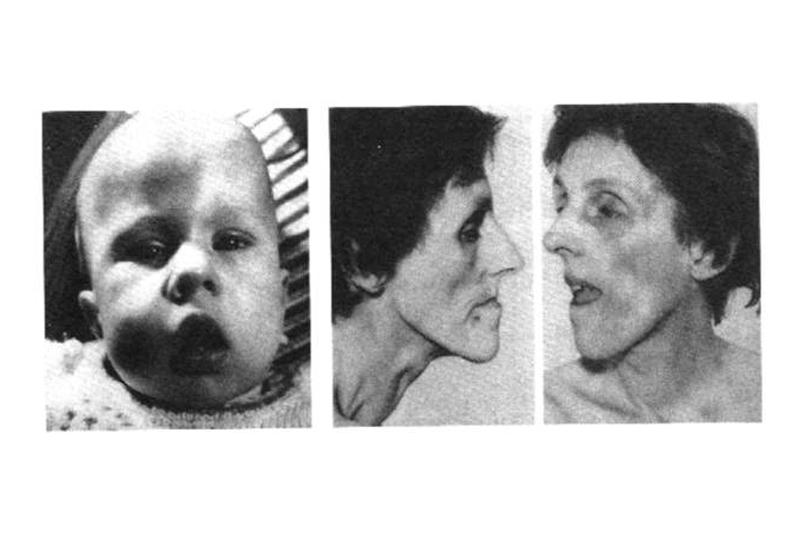
При обычном клиническом течении болезни Штейнерта, за исключением тяжелых младенческих форм, при рождении патологии не выявляется или атрофия и гипотония мимических мышц могут служить ранними симптомами заболевания. Характерен внешний вид лица ребенка: инверсия и V-образная форма верхней губы, худые щеки и фестончатые, впалые височные мышцы. Голова может быть узкой, нёбо высокое готическое в связи со слабостью височных и крыловидных мышц в позднем фетальном периоде, не оказывающих достаточного давления сбоку на растущие кости черепа и лицевого скелета.
В первые несколько лет болезни Штейнерта мышечная слабость умеренно выражена. Затем отмечаются прогрессирующая атрофия дистальных мышечных групп, особенно мышц кистей рук, уплощение тенара и гипотенара; атрофия дорсальных межкостных мышц приводит к появлению выраженных углублений между пальцами. Мышцы дорсальной поверхности предплечья и передней поверхности нижних конечностей также атрофируются. Язык тонкий и атрофированный.
Атрофия грудиноключично-сосцевидной мышцы обусловливает формирование тонкой длинной шеи цилиндрической формы. В конечном итоге проксимальные мышцы также подвергаются атрофии и формируются крыловидные лопатки. Появляются и прогрессируют затруднения при подъеме по лестнице и симптом Го-верса. Сухожильные рефлексы обычно сохранены. Дистальная мышечная атрофия — исключение из общего правила: для миопатии характерна проксимальная мышечная атрофия, для невропатии — дистальная.
Мышечная атрофия и слабость при миотонической дистрофии медленно прогрессируют в детском и подростковом возрасте, а также у взрослых. Пациенты с миотонической дистрофией редко теряют способность к самостоятельной ходьбе, даже в позднем возрасте, хотя возможна потребность в ортопедических приспособлениях (шины, фиксаторы) для стабилизации голеностопных суставов.

Миотония — характерный признак заболевания, редко встречающийся при других формах миопатии, отсутствует у детей грудного возраста и обычно не проявляется клинически и при ЭМГ примерно до 5 лет. В редких случаях миотония может возникать уже в возрасте 3 лет. Миотония — это очень медленное расслабление мышцы после сокращения независимо от того, было ли мышечное сокращение произвольным или индуцированным при попытке вызвать сухожильные рефлексы или при электростимуляции.
При осмотре можно продемонстрировать миотонию, попросив пациента крепко сжать руки в кулак и затем быстро разжать руки. Миотонию можно индуцировать при ударе неврологическим молоточком в области тенара, а также выявить, наблюдая непроизвольное приведение большого пальца. Миотонию можно продемонстрировать при перкуссии спинки языка краем шпателя — при этом на языке появляется медленно исчезающий желобок. Тяжесть миотонии не всегда коррелирует со степенью мышечной слабости, и наиболее слабые мышцы часто дают только минимально выраженную миотоническую реакции. Миотония — это неболезненный мышечный спазм. Миалгия нехарактерна для миотонической дистрофии.
Речь пациентов с миотонической дистрофией часто характеризуется плохой артикуляцией и нечеткостью вследствие поражения мышц лица, языка и глотки. Иногда возникает затруднение при глотании. У детей с тяжелым течением заболевания существует риск аспирационной пневмонии. В некоторых случаях может возникать неполная наружная офтальмоплегия в результате слабости наружных мышц глаз.
Поражение гладких мышц ЖКТ приводит к медленному опорожнению желудка, слабой перистальтике и запору. У некоторых детей выявляется энкопрез в сочетании со слабостью анального сфинктера. У женщин с миотонической дистрофией во время родов возникают неэффективные или патологические сокращения матки.
Поражение сердца чаще проявляется блокадой в проводящей системе волокон Пуркинье или аритмией, чем кардиомиопатией, в отличие от большинства других мышечных дистрофий.
Эндокринные нарушения отличаются разнообразным характером и могут возникать на любом этапе развития заболевания, поэтому повторная оценка эндокринного статуса должна проводиться ежегодно. Гипотиреоз встречается часто, гипертиреоз возникает в редких случаях. Нарушение функции коры надпочечников может привести к острой надпочечниковой недостаточности даже в грудном возрасте. Сахарный диабет часто встречается у пациентов с миотонической дистрофией; у некоторых детей нарушается высвобождение, а не синтез инсулина.
Возможно чрезмерно раннее или, чаще, позднее наступление пубертата. Атрофия яичек и дефицит тестостерона — частые признаки у взрослых пациентов, они служат причиной мужского бесплодия. Атрофия яичников встречается редко. Для мужчин также характерно облысение в лобной области, которое часто начинается в подростковом возрасте.
Для миотонической дистрофии характерен иммунодефицит. В плазме часто выявляется снижение уровня IgG.
Часто формируется катаракта. Она может быть врожденной либо начинает развиваться в любом возрасте у детей или взрослых пациентов. Ранние признаки катаракты выявляются только при исследовании с помощью щелевой лампы, рекомендуется периодический осмотр офтальмолога. В детском возрасте часто нарушаются зрительные ВП, не связанные с развитием катаракты и обычно не сопровождающиеся нарушением зрения.
Примерно у 50 % пациентов выявляются интеллектуальные нарушения, но тяжелая умственная отсталость нехарактерна. У остальных пациентов интеллект соответствует среднему уровню и иногда выше среднего уровня. Эпилепсия нехарактерна при миотонической дистрофии.
Тяжелая неонатальная форма миотонической дистрофии развивается у немногих детей, рожденных от матерей с миотонической дистрофией. Выявляется косолапость или более обширная и распространенная контрактура многих суставов, при этом могут поражаться все конечности и даже шейный отдел позвоночника. При рождении генерализованная гипотония и слабость. Выражена атрофия мышц лица. В некоторых случаях необходимы зондовое питание или ИВЛ в связи со слабостью Дыхательных мышц или развитием апноэ. Возможно одно- либо двустороннее нарушение функции диафрагмы.
Живот вздут в результате скопления газов в желудке и кишечнике в связи с нарушением перистальтики, обусловленном слабостью гладких мышц. Вздутие живота способствует еще большему усугублению дыхательных нарушений. Запор может представлять большую проблему для пациента. Около 75 % детей с тяжелой формой заболевания умирают на первом году жизни.
Дистрофическая миотония Россолимо-Штейнерта-Куршмана — это заболевание наследственного характера, прогрессирование которого происходит чрезвычайно медленными темпами. Данное нарушение наследуется только по аутосомно-доминантному типу, что происходит крайне редко — частота случаев составляет от 2 пациентов до 5 на 100 000 населения.
- Причины развития миотонии Россолимо-Штейнерта-Куршмана
- Симптомы и признаки классической миотонии Россолимо-Штейнерта-Куршмана
- Симптомы миотонии Россолимо-Штейнерта-Куршмана врожденного типа
- Диагностика миотонии Россолимо-Штейнерта-Куршмана
- Лечение миотонии Россолимо-Штейнерта-Куршмана
Заболевание является мультисистемным и характеризуется вариабельной пенетрантностью патологического гена. Сопутствующими признаками являются аритмия, патологии эндокринной системы, ухудшение интеллекта. Заболевание классифицируется на два типа: врожденный и классический.
Причины развития миотонии Россолимо-Штейнерта-Куршмана
Миотония Россолимо-Штейнерта-Куршмана — это всегда генетически передающийся порок. Ключевой причиной его развития считается нарушение в гене DMPK, размещенном в девятнадцатой хромосоме. Тяжесть течения заболевания прямо пропорциональна количеству тринуклеотидных CTG. В норме этот показатель приблизительно равен 5-37, при 50-80 наблюдается мягкая форма заболевания, при 100-500 — поздняя тяжелая форма.
Под влиянием нарушения DMPK-гена в организме происходит изменение миотонинпротеинкиназы — белка, локализующегося в скелетной и мышечной ткани, в миокарде, ЦНС и др., что провоцирует появление миотонических спазмов, сочетающихся с атрофией мышц лица, шеи и конечностей в дистальных зонах. По сути, происходит гипертрофия одних волокон мышц с одновременной атрофией других. В результате часть этих волокон замещается либо другими волокнами, либо жировыми или соединительными тканями.
Симптомы и признаки классической миотонии Россолимо-Штейнерта-Куршмана
Самые первые признаки миотонии могут проявляться уже в возрасте 6-7 лет, но чаще всего становятся хорошо заметны в подростковом и молодом возрасте от 10 до 20 лет. В перечне данных признаков присутствует миопатия, катаракта, поражения ЦНС и сердечно-сосудистой системы, эндокринные нарушения.
Основной симптом — наличие мышечных спазмов, при которых главным образом затрагивается жевательная лицевая мышца и мышца-сгибатель кисти. Также специалисты всегда отмечают атрофию различных мышц, включая дистальные отделы всех конечностей, височные, грудино-ключично-сосцевидные, мимические. Наблюдается миопатический парез гортани с затруднением дыхания, изменениями голоса, ухудшением вентиляции легких, патологиями сна и аспирационной и застойной пневмонией. Из-за изменений в мышечных тканях и преждевременного угасания рефлексов сухожилий у большинства пациентов происходит изменение походки.
Примерно в 50% случаев больные жалуются на такой симптом, как аритмия, наряду с которой могут наблюдаться блокада ножек пучка Гиса, а также гипертрофия левого сердечного желудочка.
Со стороны СНС заболевание вызывает гиперсомнию, снижение интеллектуальных способностей, нередки случаи, когда у больных отмечалась легкая форма дебильности. Из-за патологий эндокринной системы, происходят нарушения половой сферы. У молодых женщин изменения состоят в проявлении гирсутизма, сбоях менструального цикла и раннем климаксе, у молодых людей — в снижении или отсутствии влечения и импотенции, крипторхизме и гипогонадизме.
У представителей обоих полов имеется общий симптом миотонии Россолимо-Штейнерта-Куршмана — это изменения структуры волос с их последующей потерей. У мужчин облысение происходит в зоне висков и лба, у женщин проявляется местное очаговое или диффузное нарушение.
Симптомы миотонии Россолимо-Штейнерта-Куршмана врожденного типа
Врожденный тип миотонии может быть заметен специалистом еще в период пребывания в материнской утробе. Такой плод не проявляет повышенной двигательной активности. Поэтому если беременная женщина замечает подобное поведение малыша в животе, ей следует в обязательном порядке пройти УЗИ в последние три месяца перед родами.
Признаки миотонии присутствуют и у новорожденных. Это гипотония мимических, жевательных мышц, дистальных зон конечностей, глазных яблок. Кроме того, нередки и расстройства дыхания. Немного позже проявляются такие симптомы, как олигофрения и задержка моторного развития. Прогрессирование этих патологий может происходить с разной скоростью. При быстрых темпах их развития смерть пациента от заболевания может постичь его уже в самом раннем возрасте.
Диагностика миотонии Россолимо-Штейнерта-Куршмана
Подозрения специалиста на данное заболевание может вызвать сочетание любых миотонических проявлений и дистрофических изменений в тканях мышц, происходящих одновременно с отставанием в интеллектуальном развитии, нарушениями деятельности эндокринной и сердечно-сосудистой системы. Наличие у больного всех этих признаков требует подтверждения диагноза в виде результатов проведения генеалогического анализа, результаты которого говорят об аутосомно-доминантном типе генетического наследования. Кроме них для точного диагностирования следует получить данные анализа ДНК. Необходимы также дополнительные исследования: электромиография, электронейрография, электрокардиограмма, анализ гормонального фона.
Выяснение точного диагноза при подозрении на наличие миотонии Россолимо-Штейнерта-Куршмана требует всестороннего изучения состояния пациента и работы практически всех его органов. Поэтому к процессу диагностирования могут быть привлечены специалисты по генетике, кардиологии, гинекологии, андрологии и эндокринологи.
При диагностировании нужно обращать внимание не только на те изменения, которые проявляются внешне и являются характерными для миотонии Россолимо-Штейнерта-Куршмана, но и на данные по биопсии мышц и электромиографическому исследованию.
Во время проведения биохимического анализа изучается мышечный фермент и его уровень в тканях, который при данном заболевании всегда повышен. Обязательным условием постановки точного диагноза является проведение антенатальной диагностики миотонии методом амниоцентеза.
Дифференциальный диагноз миотонии Россолимо-Штейнерта-Куршмана
Необходимо дифференцировать миотонию Россолимо-Штейнерта-Куршмана от прочих известных видов миотонии, которые в ряде ситуаций могут иметь схожие признаки и течение. Атрофия мышечных тканей характерна исключительно для этого заболевания и позволяет быстро отличить его от миотонии Томсена, признаком которой является гипертрофия мышц. Раннее поражение лицевых мышц и доминантное наследование отличают его от миотонии Беккера. Помимо указанных состояний, необходимо дифференцировать заболевание Россолимо-Штейнерта-Куршмана от БАС и амиотрофии Шарко-Мари-Тута.
Лечение миотонии Россолимо-Штейнерта-Куршмана
В современной медицинской практике в настоящее время нет терапии, предназначенной для полного излечения людей от миотонии Россолимо-Штейнерта-Куршмана. Пациентам, у которых обнаружено это заболевание, назначают диету, основу которой составляют продукты с минимальным уровнем содержания калия или вовсе без него. Людям, страдающим данной миотонией, не следует переохлаждаться, так как низкие температуры могут в любом момент спровоцировать спазмы.
Для уменьшения риска их внезапного появления и улучшения общего состояния больного, ему назначают следующие препараты: хинин, новокаинамид, дифенин одновременно с диакарбом. К этим медикаментам специалисты рекомендуют добавить обязательный прием анаболических стероидов: неробола, ретаболила, метиландростендиола. Необходимо также включить в список обязательных для приема веществ небольшие дозы АТФ и витамины группы В.
Указанные выше препараты оказывают хороший эффект как при классическом, так и при врожденном типе заболевания. Все перечисленные меры позволяют снизить негативные влияния заболевания на организм пациента и продлить срок его жизни, однако они никак не способны полностью избавить его от миотонии и снова сделать абсолютно здоровым человеком.
Читайте также:
