
Миопатия лечение миопатия позвоночника


Описание
Миопатия – заболевание, которое возникает вследствие нарушения в метаболизме и строении мышечной ткани, в результате чего появляется снижение силы пораженных мышц, а также происходит ограничение двигательной активности.
Миопатии относятся к группе нервно-мышечных заболеваний, для которой характерно дистрофическое поражение мышечной ткани с атрофией отдельных волокон (миофибрилл) в некоторых местах. Кроме этого, происходит замещение атрофированных миофибрилл соединительной или жировой тканью. Это приводит к утрате мышцами способности к сокращению, что обуславливает появление в клинике заболевания мышечной слабости, а также ограничение двигательной активности.
Выделяют две основные формы заболевания:
- Первичные миопатии, которые зачастую имеют наследственный характер. В свою очередь они делятся на следующие виды:
- врожденные миопатии, которые развиваются в младенческом возрасте;
- ранние детские миопатии, которые развиваются в возрасте 5-10 лет;
- юношеские миопатии, которые развиваются в подростковом возрасте.
- Вторичные миопатии, возниающие на фоне других заболеваний.
Например, на фоне эндокринных расстройств (болезнь Иценко-Кушинга, гиперальдостеронизм, гиперпаратиреоз, гипо- и гипертиреоз), хронических интоксикаций (алкоголизм, наркомания, работа на предприятиях с профессиональными вредностями), тяжелых хронических заболеваний (хроническая почечная недостаточность, хроническая печеночная недостаточность, хроническая обструктивная болезнь легких, сердечная недостаточность), а также опухолевых процессов.
Первичные формы миопатии встречаются значительно чаще, чем вторичные. Поэтому крайне важно во время планирования беременности проконсультироваться у генетика для того, чтобы выявить риск развития данной патологии у будущего ребенка.
Исходя из того, в какой части тела мышечная слабость выражена больше, выделяют следующие виды миопатии:
- проксимальная – патологический процесс располагается в тех частях конечностей, которые максимально близко расположены к туловищу (например, в случае с руками – это плечи);
- дистальная – поражаются мышцы конечностей, которые на максимальном расстоянии находятся от туловища (например, в случае с ногами – икроножные мышцы);
- смешанная – имеет симптомы проксимальной и дистальной форм болезни.
Первичные миопатии имеют неблагоприятный прогноз. Особенно, если первые симптомы проявились в раннем детстве. Также имеет значение вовлечение в процесс сердечной мышцы и дыхательной мускулатуры. В этом случае прогноз заболевания ухудшается. Наиболее благоприятный исход наблюдается при вторичных миопатиях, так как в этом случае легче достичь успех в лечении основного заболевания, вызвавшего развитие миопатии.
Симптомы
Кроме того, миопатии могут сопровождаться поражением мимических мышц. Заподозрить вовлечение в патологический процесс мимических мышц можно в том случае, когда человек не может вытянуть губы трубочкой, надуть щеки, нахмурить лоб или улыбнуться. Поражение круговой мышцы рта сопровождается развитием дизартрии за счет затруднения произношения гласных звуков. Сама по себе дизартрия обозначает расстройство речи, которое выражается в затрудненном произношении некоторых слов, отдельный звуков, слогов или в их искаженном произношении.
Поражение дыхательной мускулатуры сопровождается нарушением вентиляции легких, что приводит к возникновению застойной пневмонии. Данная форма пневмонии является одной из самых тяжелых по течению, с трудом поддается лечению и быстро приводит к развитию дыхательной недостаточности.
Также имеются данные о возможности поражения сердечной мышцы. В таком случае развивается кардиомиопатия, которая приводит к развитию сердечной недостаточности.
Диагностика
Затем врач переходит к неврологическому осмотру, в ходе которого выявляет ослабление сухожильных рефлексов, снижение мышечной силы. В отличие от невропатии, при миопатии не наблюдается нарушение чувствительности.
После осмотра пациент отправляется в лабораторию для сдачи необходимого перечня анализов. Одним из них является биохимический анализ мочи, в котором ищут креатинин для подтверждения миопатии. Наличие креатинина в моче может указывать на поражение мышц, поэтому данный показатель не лишен информативности в диагностике рассматриваемого заболевания. Далее выполняется биохимический анализ крови, в котором оценивают уровень КФК, АЛТ, ЛДГ и других ферментов.
Помимо этого, используются элетрофизиологические методы исследования: электронейрография и электромиография. Результаты электронейрографии дают информацию о состоянии периферического двигательного нейрона, что необходимо в случае, если присутствуют подозрения на наличие нейропатии. Электромиография проводится для непосредственной оценки состояния мышечной ткани. При миопатии происходит изменение мышечных потенциалов (уменьшается их амплитуда и сокращается длительность).
Самым информативным методом является морфологическое исследование мышечной ткани. Это производится с помощью биопсии мышц. Данный метод хоть и наиболее информативный, но проводится редко, лишь в тех случаях, когда предыдущие методы исследования не дали должной информации для подтверждения того или иного диагноза. При миопатии морфологическая картина образцов мышечной ткани следующая: выявляются беспорядочно разбросанные атрофированные миофибриллы (сократительные элементы мышечных волокон), гипертрофированные мышечные волокна, а также наблюдается замещение участков мышечной ткани на соединительную или жировую.
Для оценки состояния сердечной мышцы назначается консультация кардиолога, который в свою очередь отправляет пациента на ЭКГ и УЗИ сердца. При подозрении на возникновение пневмонии, как осложнение течения миопатии, выполняется рентгенография легких.
Лечение
Для лечения первичных форм миопатии не существует препаратов, которые способны устранить генетически дефекты, послужившие развитию заболевания. Лечение в таком случае носит лишь симптоматический характер. Оно направлено на ослабление симптомов заболевания и улучшение качества жизни.
С этой целью назначаются анаболические гормоны, антихолинэстеразные препараты, витаминные комплексы. Эффект данных препаратов направлен на торможение дистрофических и атрофических изменений в мышечной ткани.
Вторичные формы миопатии лучше поддаются лечению, так как терапия направлена на устранение сопутствующих заболеваний, которые послужили причиной развития миопатии. В случае грамотного подбора медикаментозных и немедикаметозных методов лечения наблюдается постепенное устранение симптомов, беспокоящих людей с миопатией на фоне эндокринных нарушений, хронических интоксикаций, тяжелых хронических заболеваний и т.д.
Вышеперечисленные методы лечения дополняются назначением физиотерапии. Например, используется электрофорез с неостигмином, ионофорез с кальцием и ультразвук.
Помимо этого, важно назначить массаж и ЛФК. Использование данных методов лечения направлено на замедление процесса дистрофического изменения мышечной ткани с последующей атрофией. Также за счет постоянной двигательной активности уменьшается процесс перерождения мышечной ткани в соединительную и жировую. Однако важно не перегрузить ослабленные мышцы, поэтому комплекс упражнений подбирается квалифицированным специалистом.
Большое внимание уделяется дыхательной гимнастике, действие которой направлено на улучшение вентиляции легких. Это необходимо для предупреждения развития осложнения в виде пневмонии, которая может возникнуть вследствие поражения дыхательной мускулатуры.
Лекарства
Цель лечения направлена на снижение атрофии мышечной ткани. В этом значительную помощь оказывают анаболические гормоны (ретаболил, неробол). Их действие выражается в стимуляции роста мышечных клеток, что необходимо для поддержания мышечной массы. За счет этого происходит увеличение показателя силы, производительности и выносливости. Однако существует и обратная сторона приема данных препаратов. При длительном приеме могут возникнуть сопутствующие эффекты в виде облысения, увеличения роста волос на лице и теле. Помимо этого, выделяют такое понятие, как маскулинизация, которое обозначает появление вторичных половых признаков мужского пола у женщин. В этом случае происходит изменение телосложения по мужскому типу, огрубевает голос, нарушается менструальный цикл, изменяется эластичность кожи, появляется угревая сыпь.
Из антихолинэстеразных препаратов применяются такие лекарственные средства, как прозерин, нейромидин. Предпочтение отдается нейромидину, так как его спектр действия намного шире прозерина. Также при длительном приеме прозерина отмечаются различные неприятные побочные эффекты, поэтому в большинстве случаев препарат назначается короткими курсами.
В свою очередь нейромидин оказывает хорошее стимулирующее действие на нервно-мышечную передачу, а также увеличивает сократительную активность мышц. Кроме этого, нейромидин обладает седативным и анальгетическим (устраняет боль) действиями.
Из витаминных препаратов используются витамины группы Е, В и С, а также никотиновая кислота.
Народные средства
Так как в большинстве случаев причиной миопатии является наследственная патология, облегчить свое состояние без медикаментозных препаратов невозможно, но с помощью самостоятельного соблюдения некоторых мер можно улучшить общее состояние.
Например, большую роль оказывает питание, поэтому важно следовать некоторым рекомендациям. Во-первых, следует внести в свой рацион больше свежих овощей и фруктов, молочные продукты, яйца, мед, орехи, крупы (особенно овсяную и гречневую). Во-вторых, важно отказаться от кофе, алкоголя, картофеля, капусты, пряных продуктов.
Для того, чтобы предотвратить ускоренную атрофию мышц, необходимо постоянно выполнять лечебную гимнастику. Но следует также помнить, что состояние мышц при миопатии далеко от состояния мышц здорового человека. Соответственно, нагрузка должна быть минимальной, чтобы не вызвать перенапряжение. С этой целью отлично подойдут занятия в бассейне.
Также не стоит забывать о важности дыхательной гимнастике. Не редкое явление при миопатии – поражение дыхательной мускулатуры, вследствие чего нарушается вентиляция легких, что в дальнейшем приводит к воспалительному процессу (развитие пневмонии). Дыхательная гимнастика направлена на улучшение газообмена в легких.
Не рекомендуется самостоятельно разрабатывать перечень упражнений, лучше обратиться к специалисту для индивидуального подбора необходимых упражнений. После обучения в рамках лечебного учреждения можно продолжить гимнастику в домашних условиях.
Миопатия это группа тяжелых заболеваний, характеризующихся потерей тонуса мышц, ослаблением, последующей их атрофией, заменой другими видами тканей.

Когда-то активный ребенок, способный уже сидеть, ходить, бегать, постепенно теряет способность двигаться. Поражаются сначала мышцы конечностей, за ними – всех остальных органов. Заболевание приводит к тяжелым последствиям.
Миопатия: виды
В группу миопатий принято включать хронические, постепенно прогрессирующие заболевания, связанные с поражением, исчезновением мышечных волокон, заменой их жировыми или соединительными тканями. Четкой классификации на сегодня не существует.
Многие исследователи выделяют патологии по области преимущественного поражения, например, лице-лопаточно-плечевую, конечностно-поясную. Другие говорят о миопатиях в зависимости от характера причин – наследственных или приобретенных. Разделяют патологии по тому, что преимущественно поражается – белки или ферменты.
Принято также выделять отдельные болезни:
- Мышечная дистрофия Дюшенна. Болезнь встречается в 0,03% случаев. Развивается у мальчиков до 5 лет. Это одна из самых тяжелых форм миопатий. В среднем к 15 годам ребенок теряет возможность двигаться и самостоятельно себя обслуживать. Патология поражает сначала мышцы нижних конечностей, затем – верхних. В некоторых случаях развивается умственная отсталость.
- Ювенильная мышечная дистрофия Эрба-Рота. Заболевание регистрируется обычно в подростковом и юношеском возрасте до 20 лет. Поражаются мышцы таза и нижних конечностей. Больного отличает походка, напоминающая утку, тонкая талия. Отмечаются крыловидные лопатки. Обследование выявляет ухудшение сухожильных рефлексов. Из положения лежа больные встают с помощью рук.
- Плече-лопаточно-лицевая форма Ландузи-Дежерина. Миопатию этой формы отличает поражение мышц лица, плечевого пояса, лопаток. Страдают мышцы-разгибатели пальцев. Обнаруживается изменение формы груди. Заболевание впервые диагностируется в возрасте от 10 до 20 лет. В отличие от многих миопатий эта форма развивается медленно и имеет относительно благоприятный прогноз.
- Дистальная миопатия (тип Веландера). Появляется после 20 лет. Связана с уменьшением объема мышечной ткани в голеностопном отделе, коленях, предплечьях, кистях рук. Инвалидизация может наступить через 10 лет, а то и позже.
- Поздняя дистрофия Беккера. Обнаруживается у детей от 5 лет и молодых людей до 20 лет. Проявляется слабостью мышц, высокой утомляемостью, заменой мускульной ткани жировой. Сначала поражаются мышцы таза, бедер, голени, затем болезнь поражает руки. Интеллект остается сохранным.

Симптомы
Наиболее ярко признаки патологии проявляются в миопатии Дюшенна. Симптомы у детей проявляются в возрасте от полутора лет. Страдают сначала мышцы ног. Ребенку становится трудно ходить, он быстро устает, падает, не может подняться по лестнице. Падения и усталость приводят к тому, что он испытывает страх и старается передвигаться сам как можно меньше. Походка начинает напоминать утиную.
Постепенно ребенок может подняться, используя только руки, но постепенно слабеют и они. Со временем поражаются мышцы груди, сердца, дыхательных органов.
Синдром Дюшенна характеризуется постепенным исчезновением мышц, однако внешне это бывает незаметно. Наоборот, кажется, что они становятся больше. Происходит это из-за замены мышечной ткани жировой.
Заболевание приводит к нарушению скелета. Уменьшается объем движения суставов, искривляется позвоночник, деформируется стопа, пальцы.
Поражение мышц сердца ведет к появлению одышки, аритмии, нестабильности давления. Дыхание становится поверхностным.
Причины
В основе развития миопатий лежит генная мутация. В миодистрофии Дюшенна ведущую роль играет изменение гена, связанного с синтезом белка дистрофина, расположенного в X-хромосоме. До 70% случаев возникновения патологии происходит в результате наследования мутированного гена от матери. Оставшиеся 30% связаны с новыми патологическими изменениями, происходящими в яйцеклетке. Такие же причины вызывают патологию Беккера. Однако миопатия Дюшенна вызвана полной блокировкой выработки дистрофина.
Миопатией названа целая группа заболеваний мышечной ткани, преимущественно – скелетной мускулатуры, которая характеризуется стойким и прогрессирующим обменом веществ в ней. В результате этих нарушений мышцы постепенно теряют свою функцию, вплоть до полного поражения — у человека развивается вначале слабость мышц, вплоть до полного ограничения подвижности и невозможности вести активную жизнь.
Как проявляется миопатия на ранних и поздних стадиях заболевания?
Заболевание миопатия развивается постепенно, и на первых стадиях симптомы болезни могут быть стерты. Слабость в мышцах человек может списывать на усталость, последствия других заболеваний и т.д.
В самом начале развития миопатии врачи также могут неверно истолковать симптомы и ставить совершенно другие диагнозы.

- Слабость в мышцах, которая носит постоянный характер. После отдыха слабость остается, либо может уменьшаться, но – незначительно.
- Дальнейшее развитие миопатии характеризуется истончением, или атрофией, мышц – они становятся малоподвижными и тонкими. В результате заболевания могут страдать, как отдельные мышцы или группы мышц (например, только мышцы бедра или мышцы верхнегрудного отдела), так и все мышцы тела одновременно.
- Мышечный тонус сильно снижен – мышцы могут быть очень вялыми, появляется их дряблость.
- Вследствие слабости мышечного корсета в результате миопатии у человека возникает искривление позвоночного столба – это может быть кифоз, лордоз, сколиоз, которые со временем прогрессируют.
- На фоне истонченных мышц на отдельных участках тела, мышцы с нормальной функцией выглядят увеличенными – наблюдаются так называемые псевдогипертрофии.
- Сбор анамнеза.
- Неврологический осмотр и оценка мышечного тонуса.
- Оценка походки, рефлексов конечностей.
- Диагностика деформаций костей скелета, особенно – позвоночника.
- Лабораторные исследования – анализ крови на уровень креатинкиназы и гормонов щитовидной железы.
- Биопсия мышц – гистологическое исследование на факт дистрофии мышечного волокна.
- Генетические исследования с целью выявить наследственные факторы развития заболевания.
Причины возникновения миопатии
Среди основных факторов, которые являются причиной возникновения миопатий, известны следующие:
- Наследственная природа миопатии.
- Генетический дефект — вследствие недостаточности одного из важных ферментов, обеспечивающих нормальный обмен веществ в мышцах.
- Нарушения гормональной системы – например, тиреотоксикоз.
- Различные системные заболевания и патологии соединительной ткани– например, склеродермия.
По причинам возникновения заболевания различаются миопатии первичные и вторичные:
Первичные миопатии – это самостоятельно возникающие заболевания (чаще всего — наследственные).
К этой группе относятся:
- Врожденные миопатии (малыш рождается слабым, не способен громко кричать и хорошо брать грудь).
- Ранние детские миопатии (проявляются в возрасте 5-10 лет).
- Юношеские миопатии – возникают в пубертатном периоде.
Вторичные миопатии являются следствием и симптомом других заболеваний или состояний – например, интоксикации, нарушениях эндокринной системы).
По степени слабости мышц различают:
- Преимущественно проксимальные миопатии– проявляются слабостью мышц, которые на конечностях находятся ближе к туловищу (бедро, плечи).
- Дистальные миопатии – слабость наблюдается мышцах, расположенных дальше от тела (икроножные, предплечья и кисти рук).
- Смешанные миопатии, которые сочетают в себе дистальные и проксимальные миопатии.
- Дыхательная недостаточность, когда заболевание затрагивает дыхательную мускулатуру.
- Ограничение или полная потеря способности двигаться.
- Застойная пневмония, что является следствием малоподвижности и поражения дыхательной мускулатуры.
- Искривления позвоночника, межпозвонковые грыжи.
- Парезы, параличи.
Современные методы лечения
Наследственная миопатия не поддается излечению – возможно только ослабить основные симптомы и прогрессирование заболевания. Как правило, для поддержания позвоночника больному назначают ортезы. При миопатии показаны специальная лечебная гимнастика, дыхательная гимнастика.
При миопатии эндокринной природы — при гиперфункции щитовидной железы — больному назначается лечение антагонистами тиреоидных гормонов – тиреостатиками.
Вторичная миопатия вследствие заболеваний соединительной ткани (склеродермия) нуждается в лечении цитостатиков и стероидных гормонов.
При всех видах миопатии больным назначается белково-минеральная диета, из процедур наиболее эффективными являются массаж, растирания пораженных участков, бальнеолечение, ванны.
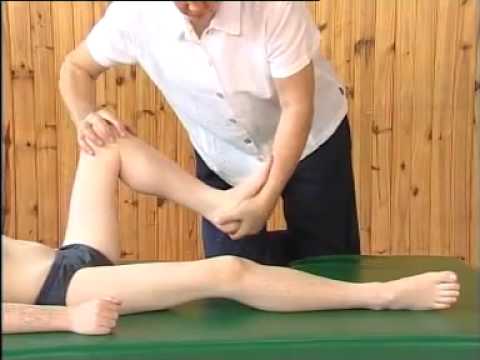
Методов оперативного лечения миопатии нет, потому что заболевание прогрессирует в любом случае. Единственно правильный путь – назначение больному ортопедических корсетов в самом начале заболевания для поддержания позвоночника и профилактики его искривления. Корсеты могут индивидуально разрабатываться и подбираться врачом-ортопедом.
Медицинский справочник болезней
Миопатия. Формы, причины, симптомы и лечение миопатий.
Миопатия — прогрессирующая мышечная дистрофия — сборная группа заболеваний, характеризующихся первичным дистрофическим процессом в мышечной ткани.
Причины.
Относится к наиболее часто встречающимся хроническим заболеваниям нервно-мышечного аппарата и носит наследственный характер.
Различные экзогенные вредности (травмы, инфекция, интоксикация) могут выявить имеющуюся патологию или вызвать ухудшение текущего процесса. Для установления семейного характера заболевания необходим не только тщательный анамнез, но и по возможности более полный осмотр всех членов семьи с выявлением так называемых малых признаков заболевания.
Наличие спорадических случаев не исключает наследственную природу.
Следует также иметь в виду возможность фенокопий миопатии, т.е. симптоматических форм или миопатических синдромов.
Патологоанатомия.
При патологоанатомических исследованиях в нервной системе не находят характерных изменений. В редких случаях отмечаются незначительное уменьшение клеток передних рогов спинного мозга, иногда — изменения в двигательных нервных окончаниях в виде набухания миелиновой оболочки, изменения осевых цилиндров. В моторных бляшках исчезает фибриллярная структура. Грубые изменения находят в поперечнополосатых мышцах. Мышцы истончены, большая часть волокон замещена соединительной тканью и жиром. Характерна неравномерность отдельных мышечных волокон — одни волокна резко уменьшены, другие, наоборот, резко увеличены.
В поздних стадиях заболевания почти вся мышечная ткань замещена соединительной или жировой тканью. В сосудах наблюдается пролиферация адвёнтиции, сужение просвета и иногда пристеночное тромбообразование. По мере развития процесса увеличивается общая масса эндо- и перимизиальной соединительной ткани с образованием фиброзного футляра вокруг мышечных волокон и внутримышечных кровеносных сосудов.
При гистохимическом исследовании наблюдается увеличение кислых муко- полисахаридов в основном веществе мышц и коллагеновых волокнах.
Патогенез.
Патогенез миопатий до настоящего времени неясен. Первичный биохимический дефект не установлен. Наиболее значительным изменениям подвергается белковый и углеводный обмен в мышечной ткани. В последнее время высказана гипотеза о нарушении обмена циклических нуклеотидов (циклических АМФ и ГМФ), являющихся универсальными регуляторами внутриклеточного обмена и ответственными за реализацию генетической информации.
Клиническая картина.
Симптоматика миопатий характеризуется нарастающими атрофиями произвольной мускулатуры. Параллельно развитию мышечного похудания появляются и парезы, однако мышечная слабость обычно выражена меньше, чем степень атрофии.
В связи с медленным прогрессированием процесса и неравномерностью поражения отдельных мышечных групп и даже участков мышц создаются условия для относительной компенсации двигательного дефекта: больные миопатией длительное время остаются трудоспособными и могут себя обслуживать, прибегая к ряду характерных вспомогательных движений. Постепенно угасают сухожильные рефлексы. Чувствительность, координация движений не нарушаются. Тазовые функции всегда сохранены.
Для некоторых видов миопатий характерны наличие псевдогипертрофий, наклонность к концевым атрофиям и сухожильным ретракциям. Фасцикулярные подергивания отсутствуют. Механическая возбудимость мышц снижена.
Нередко наблюдаются те или иные изменения внутренных органов,
- главным образом сердца: расширение границ, глухость тонов, нарушение проводимости, подтверждаемое электрокардиограммой;
- страдает функция внешнего дыхания;
- вегетативные нарушения выражаются цианозом кистей и стоп, резко повышенной потливостью, похолоданием дистальных отделов конечностей, асимметрией кожной температуры, пульса, повышенным пиломоторным рефлексом;
- страдает микроциркуляция в мышцах конечностей;
- рентгенография трубчатых костей позволяет обнаружить дистрофические явлени я;
- при электромиографическом исследовании констатируется характерная картина — снижение амплитуды биопотенциалов при достаточной частоте, а также укорочение длительности одиночного потенциала и полифазный характер;
- при биохимических исследованиях находят нарушения в креатин-креатининовом обмене. Почти всегда в моче значительно уменьшается количество креатинина и появляется креатин. Креатиновый показатель в известной степени говорит о тяжести дистрофического процесса. Отмечается значительная аминоацидурия. При ряде форм миопатий очень рано (в преклинической или в начальной клинической стадии) можно обнаружить увеличение ферментов в сыворотке крови. В первую очередь это касается специфического для мышечной ткани фермента креатинфосфокиназы;
- увеличивается также содержание аминофераз и альдолаз . Уменьшена артериовенозная разница содержания сахара крови, увеличено содержание пировиноградной и молочной кислот в мышцах и в крови. Как правило, уменьшен уровень лимонной кислоты в крови. Имеются данные о специфичности некоторых биохимических изменений при различных типах миопатий.
Классификация миопатий.
До настоящего времени нет достаточно обоснованной и общепринятой классификации миопатий. В большинстве случаев используют классификацию, основанную на клиническом принципе.
Псевдогипертрофическая форма Дюшенна является одной из наиболее частых форм миопатий. Она характеризуется самым ранним началом заболевания — нередко с 2—5-летнего возраста, а иногда даже с первого года жизни, и наиболее злокачественным течением. В типичных случаях дети к 10—12 годам уже с трудом ходят и к 15 годам становятся полностью обездвиженными.
В первую очередь страдают мышцы проксимальных отделов нижних конечностей, тазового пояса, затем в процесс вовлекаются мышцы проксимальных отделов рук. Рано выпадают коленные рефлексы. Характерна псевдогипертрофия икроножных мышц; часто уплотнение и гипертрофия их являются первым симптомом заболевания.
Псевдогипертрофии могут наблюдаться и в других группах мышц — ягодичных, дельтовидных, иногда - в языке; отмечаются значительно выраженные ретракции, в первую очередь со стороны ахилловых сухожилий. Нередко страдает сердечная мышца. Отмечается снижение интеллекта в разной степени выраженности. Для миопатии Дюшенна очень характерен высокий уровень ферментов сыворотки крови, особенно креатинфосфокиназы. Повышенный уровень ферментов можно обнаружить и у носительниц мутантного гена.
Заболевание передается по рецессивному, сцепленному с Х-хромосомой типу. Болеют только мальчики, матери являются кондукторами. Риск заболевания сыновей матерей-носительниц 50 %; 50 % дочерей становятся носителями патологического гена. Пенетрантность высокая.
Доброкачественная псевдогипертрофическая миопатия, сцепленная с Х-хромосомой (миопатия Беккера), выделена в самостоятельную форму в связи с рядом особенностей. Начало заболевания — чаще между 12 и 25 годами, иногда раньше. Течение мягкое, прогрессирование медленное, больные многие годы сохраняют трудоспособность или самообслуживание. Интеллект всегда сохранен. В остальном клиническая картина подобна псевдогипертрофической форме Дюшенна.
Плечелопаточно-лицевая форма Ландузи — Дежерина — относительно часто встречающийся тип миопатии.
Начинается заболевание, как правило, в детском или юношеском возрасте; течение сравнительно благоприятное. Первые симптомы касаются поражения мышц лица, особенно круговой мышцы рта, или мышц плечевого пояса. К этому присоединяется слабость и похудание мышц проксимальных отделов рук, затем развивается парез дистальных отделов ног. Наблюдаются умеренные гипертрофии мышц, а затем своеобразные патологические позы из-за неравномерности атрофии различных групп мышц и ретракций. Сухожильные рефлексы долго остаются сохраненными. Может быть асимметрия поражения.
Заболевание передается по аутосомно-доминантному типу с полной пенетрантностью, страдают в равной степени мужчины и женщины. Отмечаются отчетливые различия в тяжести клинических признаков не только в разных семьях, но и у разных членов одной семьи (могут быть тяжелые, легкие и абортивные формы).
Описаны стертые формы. В сыворотке крови находят умеренное повышение ферментов. Начало заболевания очень вариабельно— от детского возраста до сравнительно зрелого, но чаще в начале второго десятилетия, что отражает название этой формы. Также изменчив характер течения — иногда мягкое, благоприятное, иногда очень злокачественное.
Большинство авторов признают аутосомно-рецессивный тип наследования заболевания. Часты спорадические формы и фенокопии. Болеют одинаково часто мужчины и женщины.
Дистальная форма миопатии встречается редко.
Характеризуется поражением мышц голеней, стоп, предплечий, кистей, постепенно процесс генерализуется. Отмечаются ретракции, концевые атрофии мышц. Заболевание начинается в сравнительно позднем возрасте — 20—25 лет; прогрессирование, как правило, медленное. Отсутствие расстройств чувствительности, нормальная скорость проведения возбуждения, повышенный уровень сывороточных ферментов отличают дистальную миопатию от невральной амиотрофии.
Тип наследственной передачи—аутосомно-доминантный с неполной пенетрантностью. Несколько чаще болеют лица мужского пола.
Лопаточно-перонеальная амиотрофия (миопатия Давиденкова) проявляется поражением дистальных мышц нижних и проксимальных отделов верхних конечностей и мышц плечевого пояса. Начинается заболевание сравнительно поздно — в 25—30 лет. Наблюдаются концевые атрофии, например в большой грудной мышце, иногда крупные фасцикулярные подергивания, раннее угнетение сухожильных рефлексов.
В ряде случаев отмечаются легкие расстройства чувствительности — дистальные парестезии, гипестезии, иногда умеренная боль. Почти никогда не бывает креатинурии.
При электромиографическом исследовании выявляются специфические изменения, отличающие эту форму от обычной миопатии и невральной амиотрофии Шарко — Мари (дизритмичные колебания в покое, снижение амплитуды, уменьшение частоты и иногда групповые спайковые разряды при активных движениях).
Таким образом, данная форма является как бы промежуточной между первичной миопатией и невральной амиотрофией. Некоторые авторы рассматривают эту форму лишь как особый вариант (скапуло-перонеальный синдром) плечелопаточно-лицевой миопатии Ландузи — Дежерина.
Редкие варианты миопатии.
Описано большое количество различных вариантов прогрессирующего мышечного поражения наследственного характера. Так, например, описаны миопатия четырехглавой мышцы бедра, миосклеротическая миопатия, мышечная дистрофия с истинными гипертрофиями, врожденная мышечная дистрофия с медленным и быстрым прогрессированием, иногда с катарактой, мышечный инфантилизм и др.
Непрогрессирующие миопатии включают группу заболеваний, отличающихся или своеобразными изменениями строения мышечных клеток, или специфическими биохимическими нарушениями. Проявляются эти состояния сравнительно рано, обычно на 1—3-м году жизни, имеют сравнительно благоприятное течение. Диагноз может быть поставлен после биопсии мышц, иногда только после электронно- микроскопического исследования.
Болезнь центрального стержня характеризуется резким снижением или полным отсутствием ферментативной активности в центральной части мышечного волокна, что выявляется при окраске препарата мышечной ткани трехвалентным хромом по Гомори.
Клиническая картина: снижение мышечного тонуса, дряблость мышц, задержка развития двигательных функций, в позднем возрасте — умеренная слабость проксимальных отделов и гипотрофия мышц.
На ЭМГ — уменьшение длительности колебаний потенциала и увеличение полифазных потенциалов. Передача по доминантному типу с неполной пенетрантностью. Часты спорадические случаи.
Немалиновая, или нитеобразная, миопатия проявляется врожденной мышечной слабостью конечностей и лица с понижением мышечного тонуса и отсутствием сухожильных рефлексов. Описаны изменения скелета, в частности деформация грудной клетки, сколиоз.
На ЭМГ — изменения, характерные для мышечного уровня поражения. При электронно-микроскопическом исследовании выявляются своеобразные нитевидные структуры под сарколеммой.
Миотувулярная миопатия клинически выражается в понижении мышечного тонуса, умеренно выраженных атрофиях мышц конечностей с наличием диффузной слабости рук, ног, туловища. Характерны также слабость мимической мускулатуры, птоз и ограничение подвижности глазных яблок, общая задержка развития двигательных функций. Состояние может быть стационарным или медленно прогрессирующим. У большинства больных выявляются те или иные костные деформации.
На ЭМГ — сочетание мышечного типа изменений с наличием спонтанной активности. Гистологически определяются мышечные волокна резко уменьшенной величины с центральным расположением ядер, напоминающие по строению эмбриональную мышечную ткань. При электронной микроскопии выявляются участки дегенеративно измененных миофибрилл, при гистохимическом исследовании обнаруживается повышение активности митохондри- альных ферментов.
Митохондриальные миопатии характеризуются увеличением числа митохондрий в мышечных волокнах или увеличением размеров митохондрий, обнаруживаемых при электронно-микроскопическом исследовании. Клинически отмечается мышечная слабость, главным образом в проксимальных отделах рук и ног, вялость, быстрая утомляемость при отсутствии особых мышечных атрофий. Прогрессирования, как правило, не отмечается.
Диагноз прогрессирующей мышечной дистрофии, как правило, не представляет больших трудностей. Атипичные формы приходится дифференцировать от сирингомиелии (переднероговая форма), начальных явлений амиотрофического склероза, спинальных амиотрофий, полимиозита и других миопатических синдромов (см.). Комплексное обследование больного с применением биохимических (определение уровня ферментов и др.), электрофизиологических (ЭМГ, определение скорости распространения возбуждения по нерву), гистологических исследований и анализ клинической картины позволяют поставить правильный диагноз.
ЛЕЧЕНИЕ.
- Воздействие на энергетической обмен в мышцах осуществляется назначением АТФ в виде монокальциевой соли по 3—6 мл в день внутримышечно в течение 30 дней.
- Показано применение витамина Е внутрь по 30—40 капель 3 раза в день или внутримышечно раствор токоферола ацетата в масле по 1—2 мл — 20 инъекций (или эревит).
- Назначают витамины группы В, гликокол, лейцин (по 1 столовой ложке 3 раза в день), глутаминовую кислоту (по 0,5—1 г 3 раза в день).
Накопленный опыт по применению анаболических гормонов не подтверждает возлагавшихся на этот вид лечения надежд.
- Рекомендуется сочетать медикаментозное лечение с физиотерапией (гальванический воротник и гальванические трусы с кальцием, солянохвойные ванны со строго индивидуальной лечебной физкультурой при средней нагрузке).
- При наличии контрактур может быть рекомендовано оперативное вмешательство на сухожилиях.
- Показан систематически проводимый легкий массаж.
Читайте также:
