
Новое лечение мышечной дистрофии дюшенна
16 февраля 2016
- 3800
- 3,1
- 0
- 6
Принцип генной терапии миодистрофии Дюшенна/Беккера. Миодистрофию Дюшенна (МДД) вызывают мутации гена дистрофина (DMD), приводящие к сдвигу рамки считывания, а более мягкую миодистрофию Беккера (МДБ) — мутации без смещения рамки считывания. Лечения этой болезни пока нет. Генная терапия поможет улучшить или даже восстановить функции мышц.
![]()
Анна Петренко

![]()
Антон Чугунов![]()
Ольга Волкова


- CRISPR/CAS
- Генная инженерия
- Медицина
Мышечная дистрофия Дюшенна — тяжелейшее Х-связанное заболевание, эффективного лечения которого до сих пор нет. В одном из последних номеров Science вышли целых три статьи об успешном тестировании на мышиных моделях технологии CRISPR/Cas9 для лечения этой болезни. Может быть, у этого подхода есть шанс добраться и до клиник?
Мышечная дистрофия Дюшенна, от которой страдает один из 3600-5000 новорожденных мальчиков, вызывается отсутствием дистрофина — белка, который соединяет цитоскелет и внеклеточный матрикс в мышечном волокне и обеспечивает его стабильность при сокращении (рис. 1). Из-за мутаций гена DMD рамка считывания при трансляции его мРНК сдвигается, и синтез белка преждевременно прекращается. Врожденная болезнь очень быстро прогрессирует: ее диагностируют в возрасте около четырех лет, а к 10 годам ребенку обычно уже нужна инвалидная коляска. Это происходит потому, что без дистрофина волокна повреждаются, и как только регенеративная способность мышечных волокон исчерпывается, они заменяются фиброзной и жировой тканями [1]. Как показывают исследования, когнитивные функции у ребенка тоже могут быть нарушены [2]. Больше 30 лет с таким заболеванием, как правило, не живут, а смерть наступает от сердечных и респираторных осложнений. Более мягкий вид миодистрофии, связанной с геном DMD, — это мышечная дистрофия Беккера, когда мутации не приводят к смещению рамки считывания [3].
Дистрофин находится на внутриклеточной поверхности сарколеммы вдоль всей длины мышечных волокон и входит в состав дистрофин-ассоциированного гликопротеинового комплекса (ДАГК, DGC). Он связывается одним концом с F-актином цитоскелета, а другим — с β-дистрогликаном, что стабилизирует волокна во время сокращения. Ген дистрофина — один из самых длинных у человека.
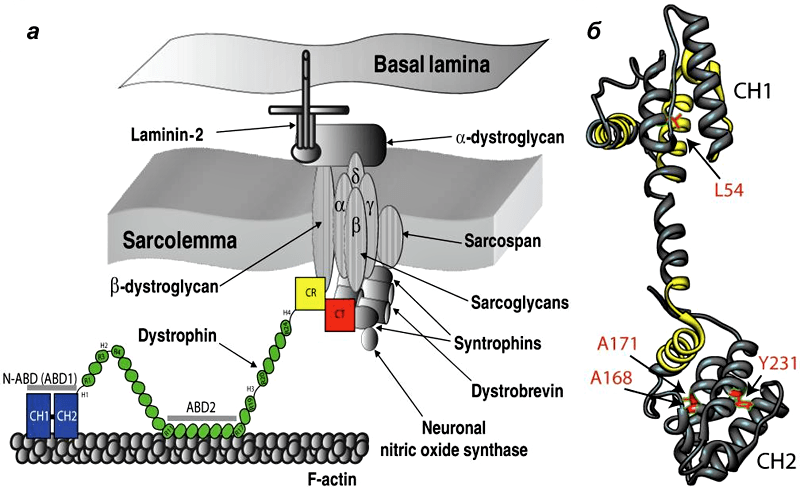
Рисунок 1. Мутации в дистрофине — причина развития миодистрофии Дюшенна. а - Дистрофин связывается с актиновыми филаментами (часть цитоскелета) через домены N-ABD и ABD2) и с ДАГК через домены CR и CT. б — Кристаллическая структура N-ABD дистрофина. Зоны связывания с актином показаны желтым, четыре хорошо изученных мутации, вызывающих заболевание, — красным.
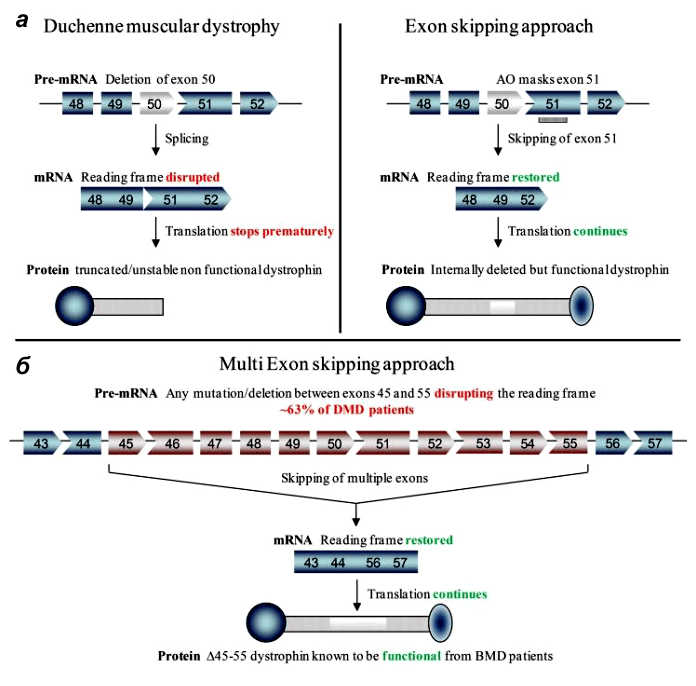
У стратегии удаления экзонов есть даже преимущества перед воссозданием полной длины гена: ее проще разработать, чем восстановить индивидуальные делеции каждого пациента [7].
Конкурирующие лаборатории: кто первым воплотит технологию в терапию для человека?
Ученые трех лабораторий успешно применили технологию пропуска экзонов in vivo на стандартном объекте — мышах — и показали, что их метод помогает восстановить рамку считывания и частично восстановить синтез дистрофина. Поскольку даже невысокий его уровень (3–15% от нормального) приносит терапевтическую пользу, результаты работ можно назвать успешными.
Группа Эрика Олсона уже не в первый раз использует метод CRISPR/Cas9 в своих работах по мышечной дистрофии Дюшенна. В 2014 году ученые исправили мутацию в зародышевой линии мышей и предотвратили развитие болезни. Однако, поскольку пренатальное редактирование генома на человеческих эмбрионах (пока?) запрещено, исследователям пришлось придумать способ постнатального применения технологии.
Группа Эми Уаджерс провела во многом похожий эксперимент [8]. После множества подготовительных этапов работы по редактированию генома и пропуску экзона на клетках и животных их опыт тоже увенчался успехом: программируемые CRISPR-комплексы в составе аденоассоциированного вируса (AAV) были доставлены с помощью локального и системного введения к дифференцированным скелетным волокнам, кардиомиоцитам и сателлитным мышечным клеткам новорожденных и взрослых мышей. Если редактирование направлено только на мышечные волокна, то эффект со временем может сойти на нет. Однако, как отмечает Уаджерс, редактирование генов в сателлитных клетках может обеспечить гораздо более длительный результат. Оно способно привести к созданию пула регенеративных клеток, несущих отредактированный ген дистрофина, и в результате обычной репарации мышц отредактированный ген окажется и в мышечных волокнах.
Терапия миодистрофии Дюшенна: старые и новые подходы
По словам Олсона, главное отличие новой стратегии с использованием вектора, вмещающего в себя компоненты для редактирования генома, от других терапевтических методов в том, что она устраняет причину болезни. А какие еще подходы разрабатывают ученые?
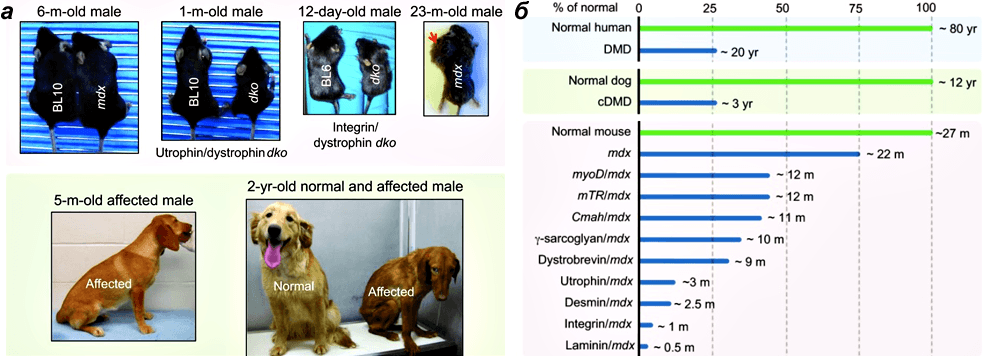
Рисунок 3. Животные модели миодистрофии Дюшенна. а — Проявления миодистрофии Дюшенна у мышей и собак. Вверху: у мышей mdx симптомы проявляются только в старости, и они склонны к образованию рабдомиосарком — опухолей мышечного происхождения. Размер мышей с нокаутами генов атрофина/дистрофина и интегрина/дистрофина значительно меньше, чем их ровесников дикого типа (BL10 и BL6). Внизу: проявления болезни у пятимесячной больной собаки. Различия между здоровой и больной двухлетними собаками. б — Сравнение продолжительности жизни здоровых и больных людей, собак и различных линий мышей.
Один из многообещающих подходов — это клеточная терапия. Хотя опыты с внутримышечной инъекцией миобластов от здоровых доноров провалились, технологии с использованием стволовых клеток и индуцированных плюрипотентных стволовых клеток (ИПСК) пока успешно испытываются на моделях не только миодистрофии Дюшенна, но и болезни Альцгеймера, Паркинсона, Хантингтона, спинальной мышечной атрофии, бокового амиотрофического склероза, аутизма и шизофрении [14–16]. Например, в 2013 году исследователи из Бостонской детской больницы (Boston Children’s Hospital’s Stem Cell Program) с помощью смеси трех малых молекул (форсколина, основного фактора роста фибробластов bFGF и ингибитора гликогенсинтазы киназы-3) перепрограммировали ИПСК из кожи пациентов с миодистрофией Дюшенна в мышечные клетки, которые затем успешно прижились у мышей. Сейчас из ИПСК получены кардиомиобласты и нейроны [2].
Другие исследования показывают, что восстановление нормального уровня синтеза оксида азота (NO), который снижается у больных из-за нарушения активности NO-синтазы (nNOS), ослабляет воспаление, повышает активность собственных стволовых клеток и реконструирует морфологию и функции скелетных мышц [3].
Уже в фазе II клинических испытаний находится препарат Givinostat — ингибитор гистондеацетилаз, который замедляет прогрессирование болезни в мышиной модели.
Такой массированный экспериментальный удар по миодистрофии Дюшенна вселяет надежду. Станет ли технология CRISPR/Cas9 ведущей в разработке терапии, которую смогут принять на вооружение клиницисты? Возможно, не за горами и публикации похожих работ по другим заболеваниям, где нужно избавиться от мутаций в одном-единственном гене? Это мы узнаем из ближайших выпусков Science (а также других почетных журналов).
Не упустить время
Милая неуклюжесть, сопровождающая малыша до трех лет, к пяти превращается в мышечную слабость. В 10-12 лет ребенок перестает ходить, к 15 — частично или полностью теряет способность двигаться самостоятельно. Всего 20 лет жизни — так мало, что в это невозможно поверить.
Врачи отделываются общими фразами, и родителям приходится искать информацию самостоятельно. Доктора, впервые столкнувшиеся с таким диагнозом, и сами зачастую не представляют, что делать.
Миопатия Дюшенна — заболевание, которое характеризуется мутацией в гене, отвечающем за синтез белка дистрофин в мышечных волокнах, что приводит к нарушению их строения. Носители мутаций — женщины, которые сами миопатией Дюшенна не болеют, но с 50% вероятностью передают ее своим потомкам. У мальчиков при этом развивается мышечная дистрофия, а девочки, в свою очередь, становятся носителями мутации. В некоторых случаях заболевание не является результатом передачи по наследству, а возникает спонтанно.
В России гигантский провал в данной области научных исследований. Специалистов по нейромышечным патологиям крайне мало, попасть к ним сложно. Только представьте, как много значит один год для прогрессирующего заболевания! А именно столько в среднем длится очередь в федеральное медучреждение.
Обычно сначала родители из-за трудностей при ходьбе обращаются к ортопедам, которые могут просто не знать об этом заболевании. Как показывает практика российских поликлиник, даже неврологи во многих случаях ошибаются с диагнозом. Тем более, что у 30% мальчиков миопатия сопровождается задержкой умственного развития.
Судя по частоте заболевания, в России должно быть около 4000 детей с дистрофией Дюшенна. На учете в московском детском нервно-мышечном центре — в 10 раз меньше. Где все остальные пациенты?
В этом-то и кроется самая большая проблема отечественной медицины. Невролог, наконец-то правильно поставивший диагноз, вольно или невольно убеждает родителей, что миопатия Дюшенна не лечится, быстро прогрессирует и всегда приводит к смерти. В результате — родители просто опускают руки, никуда не едут и ни к кому не обращаются, пытаясь просто выжить в поставленных условиях.
Правда ли, что ситуация настолько безвыходна? Неужели нет надежды на то, что ребенку действительно помогут? Разве неизлечимое заболевание — это повод отказаться от поисков наиболее эффективного лечения?
Учиться и работать с дистрофией Дюшенна — реально
Будем честны. Пока чуда мгновенного излечения ждать не стоит, кто бы вам что ни обещал. Однако продлить жизнь ребенка и в разы улучшить ее качество — возможно уже сейчас! Велик шанс, что за те годы, что вы ему подарите, ученые наконец-то найдут средство остановить или повернуть вспять развитие заболевания.
Центр комплексного лечения нейромышечных нарушений Детской больницы Цинциннати (Comprehensive Neuromuscular Center, Цинциннати, США) доказывает это каждый день:
Здесь регулярно проводят клинические испытания новых лекарств и методов, что позволило достичь отличных результатов в лечении мышечной миопатии Дюшенна.
На данный момент в Центре наблюдаются около 600 детей с этим заболеванием. Сюда приезжают со всего мира, чтобы получить современное лечение и эмоциональную поддержку. Медицинская команда оказывает своим пациентам и их семьям долгосрочную помощь.
Наблюдения за пациентами 13-16 лет, которые выполняли все назначения врачей Центра, подтверждают: к этому возрасту 40% из них могут самостоятельно встать с пола, 50% — пройти 10 метров без посторонней помощи. Тогда как без лечения ходьба становится абсолютно невозможной уже к 12-13 годам.
Все специалисты собраны в одном учреждении. Совместная работа неврологов, кардиологов, пульмонологов, эндокринологов, генетиков, физиотерапевтов, диетологов приводит к тому, что средняя продолжительность жизни людей с миопатией Дюшенна увеличилась на 10 лет.
Благодаря комплексному лечению и постоянному наблюдению вместо 20 лет пациенты могут жить 30 и более. Соответственно, и основные двигательные функции тоже сохраняются намного дольше.
А это значит, что дети могут успешно получить профессиональное образование и даже работать. Такие примеры среди пациентов Центра уже есть!
Эффективные методики лечения миопатии Дюшенна от Comprehensive Neuromuscular Center
Центр комплексного лечения нейромышечных нарушений работает по продуманной схеме, которая делает лечение максимально комфортным для пациента и его законных представителей (родителей или опекунов).
Преимущества лечения миодистрофии Дюшенна в Comprehensive Neuromuscular Center:
- Специализация на нейромышечных нарушениях. В Центр обращаются пациенты с миопатиями Дюшенна и Беккера, спинальной мышечной атрофией (СМА), врожденным миастеническим синдромом, наследственной атаксией Фридрейха, другими патологиями нервной и мышечной системы.
- Пациенты Центра первыми получают максимальный доступ к новым научным открытиям и достижениям, к самым последним методикам и фармацевтическим средствам. А еще — участвуют в клинических исследованиях.
- По результатам обследования лечащий врач составляет индивидуальный план в зависимости от текущего состояния ребенка и сопутствующих заболеваний. Учитывается даже образ жизни семьи в целом — все для того, чтобы предоставить наилучшую помощь.
- Управление побочными действиями. Состояние ребенка постоянно контролируется с тем, чтобы своевременно корректировать количество и вид принимаемых препаратов.
- Всесторонний амбулаторный и стационарный уход, которые выражаются в постоянной заботе. 100% ведение пациента в течение всей жизни вплоть до консультирования по вопросам качества жизни, обучения и адаптации в обществе.
Лечение миодистрофии Дюшенна будет намного результативнее, если в семье поддерживается благоприятная психологическая обстановка. Поэтому в заключение мы подготовили несколько советов, которые помогут вам сохранять спокойствие и уверенность, а ребенку — стать счастливее.
- Не скрывайте от ребенка диагноз, правдиво и понятно рассказывайте ему о заболевании.
- Помогите вашему ребенку понять, что он может и должен быть активным участником процесса лечения. Правильно питаясь и выполняя рекомендованные физические упражнения, пациент и сам положительно влияет на ход течения болезни.
- Помните, что ваш ребенок — это личность, состоящая из многих аспектов. И миопатия Дюшенна — лишь один из них, причем не самый главный.
- Обращайте внимание на то, что ребенок еще может сделать. Не зацикливайтесь на потерянных умениях.
- Решайте только актуальные проблемы. Не думайте о том, как ребенок будет чувствовать себя через месяц или через год.
- Воспитывайте ребенка так же, как здорового. Старайтесь избежать чрезмерной опеки, предоставьте ему право быть независимым в доступных для него сферах.
- Просите о помощи близких, друзей, врачей тогда, когда вам это нужно.
- По возможности ходите вместе в кино, ездите в отпуска, развлекайтесь — дарите себе и ребенку радостные моменты, наполненные позитивными эмоциями.
- Нужна подробная консультация по нейромышечным нарушениям? Отправьте запрос прямо с сайта и узнайте, как связаться со специалистами Центра комплексного лечения нейромышечных нарушений Детской больницы Цинциннати.
Автор Ольга Тонкушина
- 3.74
- 1
- 2
- 3
- 4
- 5

Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
- Причины мышечной дистрофии Дюшенна
- Симптомы мышечной дистрофии Дюшенна
- Диагностика мышечной дистрофии Дюшенна
- Лечение мышечной дистрофии Дюшенна
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Причины мышечной дистрофии Дюшенна
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы мышечной дистрофии Дюшенна
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика мышечной дистрофии Дюшенна
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК. При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
Лечение мышечной дистрофии Дюшенна
Терапия, применяемая в клинической практике, пока включает лишь необходимые симптоматические мероприятия. Для улучшения метаболизма мышечной ткани возможно назначение анаболических стероидов (метандиенона, нандролона деканоата), АТФ, актопротекторов (этилтиобензимидазола); для облегчения нервно-мышечной передачи — неостигмина. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия.
Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Поиск эффективных способов лечения дистрофии Дюшенна — задача, над решением которой трудятся сегодня специалисты в области неврологии, биохимии, генной инженерии. Из перспективных разработок в этом направлении можно выделить лечение стволовыми клетками, активацию гена утрофина, являющегося наиболее близким аналогом дистрофина, технологию пропуска экзонов.
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.

Мышечная дистрофия Дюшенна – это генетическое заболевание, связанное с нарушением строения мышечных волокон. Мышечные волокна при этой болезни, в конце концов, распадаются, и теряется способность к передвижению. Мышечная дистрофия Дюшенна передается сцеплено с полом, болеют лица мужского пола. Проявляет себя уже в детском возрасте. Помимо мышечных нарушений, заболевание приводит к скелетным деформациям, может сопровождаться дыхательной и сердечной недостаточностью, умственными и эндокринными нарушениями. Радикального способа лечения, позволяющего искоренить болезнь, пока нет. Все существующие меры являются лишь симптоматическими. Довольно редко больным удается пережить рубеж 30 лет. Эта статья посвящена причинам, симптомам, диагностике и лечению мышечной дистрофии Дюшенна.
Заболевание впервые описано в 1861 году (по другим данным – 1868 году) французским невропатологом и носит его имя. Встречается не так уж и редко: 1 случай на 3500 новорожденных детей. Из всех известных медицине мышечных дистрофий является наиболее распространенной.
Причина заболевания

В основе мышечной дистрофии Дюшенна лежит генетический дефект половой Х хромосомы.
Один из участков Х хромосомы содержит ген, кодирующий производство в организме особого мышечного белка под названием дистрофин. Белок дистрофин составляет основу мышечных волокон (миофибрилл) на микроскопическом уровне. Функция дистрофина заключается в поддержании клеточного скелета, в обеспечении способности миофибрилл к многократным актам сокращения и расслабления. При мышечной дистрофии Дюшенна этот белок либо отсутствует вообще, либо синтезируется дефектным. Уровень нормального дистрофина не превышает 3%. Это приводит к разрушению мышечных волокон. Мышцы перерождаются и заменяются жировой и соединительной тканью. Естественно, что при этом утрачивается двигательный компонент человеческой деятельности.
Симптомы заболевания
Мышечная дистрофия Дюшенна всегда заявляет о себе до 5-летнего возраста. Наиболее часто первые симптомы возникают еще до 3-х лет. Все патологические проявления заболевания можно разделить на несколько групп (в зависимости от характера изменений):
- поражение скелетной мускулатуры;
- деформации скелета;
- поражение сердечной мышцы;
- умственные нарушения;
- эндокринные расстройства.

Поражение мышечной ткани является основным проявлением заболевания. Оно становится причиной генерализованной мышечной слабости. Начальные симптомы подкрадываются незаметно.
Рождаются дети без особых отклонений. Однако их двигательное развитие отстает в темпах по сравнению со сверстниками. Такие дети менее активные и подвижные в двигательном плане. Пока ребенок совсем мал, это часто связывают с особенностями темперамента и не обращают внимания на начальные изменения.
Явные признаки возникают с началом ходьбы. Дети часто падают и передвигаются на пальцах (на носочках). Следует отметить, что эти нарушения интерпретируют не при первых шагах ребенка, потому что прямохождение вначале для всех детей сопряжено с падениями и неуклюжестью. Когда большинство ровесников уже вполне уверенно передвигается, мальчики с мышечной дистрофией Дюшенна упорно продолжают падать.
Когда ребенок научится разговаривать, он начинает жаловаться на слабость и быструю утомляемость, непереносимость физических нагрузок. Бег, лазанье, прыжки и другие любимые виды активной деятельности детей для ребенка с мышечной дистрофией Дюшенна не привлекательны.
Походка таких детей напоминает утиную: они как бы переваливаются с ноги на ногу.
Мышечная дистрофия Дюшенна имеет восходящий тип мышечной слабости. Это означает, что сначала слабость проявляет себя в ногах, затем распространяется на таз и туловище, потом на плечи, шею и, в конце концов, на руки, дыхательную мускулатуру и голову.
Несмотря на то, что при этом заболевании мышечные волокна подвергаются разрушению и развитию атрофии, внешне некоторые мышцы могут выглядеть вполне нормальными или даже накаченными. Развивается так называемая псевдогипертрофия мышц. Чаще всего этот процесс заметен в икроножных, ягодичных и дельтовидных мышцах, мышцах языка. Место распавшихся мышечных волокон занимает жировая ткань, именно поэтому создается эффект хорошей развитости мускулатуры, что на проверку оказывается совсем не так.
Разрушение мышц сопровождается развитием мышечных контрактур и укорочением сухожилий (хорошо заметно на примере ахиллового сухожилия).
Сухожильные рефлексы (коленный, ахиллов, с бицепса, с трицепса и так далее) постепенно снижаются. Мышцы на ощупь плотные, но безболезненные. Мышечный тонус обычно снижается.
Постепенное прогрессирование мышечной слабости приводит к тому, что к 10-12 годам многие дети утрачивают способность самостоятельно передвигаться и нуждаются в инвалидном кресле. Способность стоять сохраняется, в среднем, до 16 лет.
Отдельно следует сказать о вовлечении в патологический процесс дыхательной мускулатуры. Это наблюдается после подросткового периода. Слабость диафрагмы и других мышц, участвующих в акте дыхания, приводит к постепенному снижению жизненной емкости легких и объемов вентиляции. По ночам это особенно заметно (появляются приступы удушья), поэтому у детей могут возникать страхи перед сном. Формируется дыхательная недостаточность, которая усугубляет течение интеркуррентных инфекций.

Это сопутствующие мышечным изменениям симптомы. У детей постепенно формируются усиление поясничного изгиба (лордоза), искривление грудного отдела позвоночника в сторону (сколиоз) и сутулость (кифоз), меняется форма стопы. Со временем развивается диффузный остеопороз. Эти симптомы еще больше способствуют ухудшению двигательных нарушений.
Является обязательным симптомом мышечной дистрофии Дюшенна. У больных развивается кардиомиопатия (гипертрофическая или дилатационная). Клинически это проявляет себя нарушениями сердечного ритма, перепадами артериального давления. Границы сердца увеличиваются, однако такое большое сердце имеет малые функциональные возможности. В конце концов, формируется сердечная недостаточность. Сочетание выраженной сердечной недостаточности с дыхательными расстройствами на фоне присоединившейся инфекции может быть причиной смертельного исхода у больных с мышечной дистрофией Дюшенна.
Это не обязательный, но возможный признак заболевания. Он связан с дефицитом особой формы дистрофина – аподистрофином, содержащимся в головном мозге. Нарушения интеллекта варьируются от незначительных до степени идиотии. При этом выраженность умственных нарушений никоим образом не связана со степенью мышечных расстройств. Социальная дезадаптация из-за невозможности свободно передвигаться и посещать детские учреждения (садики, школы) способствует усугублению когнитивных расстройств.
Встречаются у 30-50% больных. Могут быть довольно разнообразными, но чаще всего это ожирение с преимущественным отложением жира в области молочных желез, бедер, ягодиц, плечевого пояса, недоразвитие (или нарушение функции) половых органов. Больные часто имеют низкий рост.
Мышечная дистрофия Дюшенна неуклонно прогрессирует. К 15-20 годам почти все больные не в состоянии себя обслуживать самостоятельно ввиду обездвиженности. В конце концов, присоединяются бактериальные инфекции (органов дыхания и мочевыделительной сферы, инфицированные пролежни при недостаточном уходе), которые на фоне сердечной и дыхательной недостаточности приводят к смертельному исходу. Мало кто из больных переживает рубеж в 30 лет.
Диагностика

Диагностика мышечной дистрофии Дюшенна основывается на нескольких видах исследований, основным из которых является генетический тест (ДНК-диагностика).
Только обнаружение дефекта Х хромосомы в том участке, который отвечает за синтез дистрофина, достоверно подтверждает диагноз. До проведения такого анализа диагноз является предварительным.
Из других методов исследования могут применяться:
- определение активности креатинфосфокиназы (КФК). Этот фермент отражает гибель мышечных волокон. Его концентрация при мышечной дистрофии Дюшенна превышает норму в десятки и сотни раз до 5-летнего возраста. Позже уровень фермента постепенно снижается, потому что часть мышечных волокон уже необратимо разрушена;
- электромиография. Этот метод позволяет подтвердить тот факт, что в основе заболевания лежат первичные изменения мышц, а нервные проводники при этом совершенно интактны;
- биопсия мышц. С ее помощью определяют содержание белка дистрофина в мышце. Однако в связи с усовершенствованием генетической диагностики в последние десятилетия эта травматичная процедура отошла на второй план;
- дыхательные пробы (исследование жизненной емкости легких), ЭКГ, УЗИ сердца. Эти методы не используются для установления диагноза, но необходимы для выявления патологических изменений со стороны дыхательной и сердечно-сосудистой систем для того, чтобы скорригировать имеющиеся нарушения.
Выявление больного ребенка в семье означает, что в генотипе матери имеется патологическая Х хромосома. В редких случаях мать может быть здоровой, если мутация возникла у ребенка случайно. Наличие дефектной Х хромосомы несет в себе риск для последующих беременностей. Поэтому такие семьи должен консультировать генетик. При наступлении повторных беременностей родителям предлагают пренатальную диагностику, то есть исследование генотипа еще не родившегося ребенка с целью исключения наследственных заболеваний, в том числе мышечной дистрофии Дюшенна.
Для исследования понадобятся клетки плода, которые получают с помощью различных процедур на разных сроках беременности (например, биопсия хориона, амниоцентез и другие). И хотя эти медицинские манипуляции несут в себе определенный риск для беременности, они позволяют точно ответить на вопрос: имеется ли у плода генетическое заболевание.
Лечение
Мышечная дистрофия Дюшенна является в настоящее время неизлечимым заболеванием. Можно помочь ребенку (взрослому) продлить время двигательной активности, с помощью различных способов поддерживая мышечную силу, компенсируя изменения со стороны сердечно-сосудистой и дыхательной систем.
Несмотря на это, прогнозы ученых в отношении полного излечения от этого заболевания довольно оптимистичны, поскольку уже сделаны первые шаги в этом направлении.
В настоящее время, для лечения мышечной дистрофии Дюшенна из медикаментозных препаратов используют:
- стероиды (при регулярном применении они позволяют уменьшить мышечную слабость);
- β-2-адреномиметики (также временно придают силу мышцам, но не замедляют прогрессирование заболевания).
Применение β-2-адреномиметиков (Альбутерол, Формотерол) не имеет статистически достоверного признания, поскольку существует небольшой опыт их использования при данной патологии. Контроль изменений состояния здоровья у группы пациентов, применявших эти препараты, проводился в течение одного года. Поэтому утверждать, что они работают более длительное время, нет возможности.
Основой лечения сегодня являются стероиды. Считается, что их использование позволяет какое-то время сохранять мышечную силу, то есть они могут затормозить прогрессирование болезни. Кроме того, доказано, что стероиды уменьшают риск возникновения сколиоза при мышечной дистрофии Дюшенна. Но все же возможности этих препаратов ограничены, и заболевание будет неуклонно прогрессировать.
Когда же начинают лечение гормонами? Считается, что оптимальным временем для начала терапии является такая фаза заболевания, когда двигательные навыки не улучшаются, но еще не ухудшаются. Обычно это бывает в возрасте 4-6 лет. Наиболее часто используются такие препараты, как Преднизолон и Дефлазакорт. Дозы назначаются индивидуально. Препараты используются, пока есть видимый клинический эффект. Когда же наступает фаза прогрессирования заболевания, то необходимость применения стероидов отпадает, и их постепенно (!) отменяют.
Из медикаментозных препаратов также при мышечной дистрофии Дюшенна применяют сердечные средства (антиаритмические, метаболические, ингибиторы ангиотензинпревращающего фермента). Они позволяют бороться с кардиологическими аспектами заболевания.
Ортопедические приспособления позволяют значительно облегчить жизнь больного. Их перечень довольно широк и разнообразен: это и различного рода вертикализаторы (помогают сохранять положение стоя), и приспособления для самостоятельного вставания, и инвалидные кресла-коляски с электрическим приводом, и специальные шины для устранения контрактур в голени (используются даже ночью), и корсеты для позвоночника, и длинные шины для ног (колено-голеностопные ортезы), и многое другое.
Когда заболевание поражает и дыхательные мышцы, и самостоятельное дыхание становится неэффективным, то возможно применение аппаратов искусственной вентиляции легких различной модификации.
И все же даже использование всех этих мер в комплексе не позволяет побороть заболевание. На сегодняшний день, есть ряд перспективных направлений исследований, которые, возможно, станут прорывом в лечении мышечной дистрофии Дюшенна. К наиболее распространенным среди них относят:
Каждая из новых разработок несет в себе надежду для больных с мышечной дистрофией Дюшенна на полное выздоровление.
Таким образом, мышечная дистрофия Дюшенна – это генетическая проблема лиц мужского пола. Болезнь характеризуется прогрессирующей мышечной слабостью из-за разрушения мышечных волокон. В настоящее время является неизлечимым заболеванием, однако многие ученые мира трудятся над созданием радикального способа борьбы с ним.
Читайте также:
