
Мышечная атрофия плечевого пояса
К этой группе относятся: псевдогипертрофический паралич Дюшена, мышечные атрофии типа Лейден-Мобиуса, Ландузи-Дежерина, Циммерлина и юношеская форма Эрба. Последние четыре формы представляют только клинические разновидности псевдогипертрофического паралича, которые характеризуются либо особым началом, либо некоторыми особенностями в состоянии атрофированных мышц.
Псевдогипертрофический паралич Дюшенна
Псевдогипертрофический паралич, как и клинические разновидности его, представляет преимущественно семейную и наследственную болезнь; он встречается у нескольких членов и иногда в нескольких поколениях одной и той же семьи. Женщины поражаются гораздо реже, чем мужчины.

Болезнь начинается большей частью в раннем детстве, между тем как мышечная атрофия типа Лейден-Мобиуса, развивается чаще у детей старшего возраста, а мышечная атрофия типа Циммерлина – в юношеском возрасте.
Большей частью первым явлением бывает расстройство походки; она становится неуверенной, ребенок быстро устает при ходьбе и часто падает. Эта слабость нижних конечностей скоро обращает на себя внимание родителей, тем более что они начинают замечать у ребенка увеличение объема мышц сперва на икрах, затем на ягодицах, которое не соответствует мышечной слабости.

По мере того, как болезнь прогрессирует, все явления усиливаются. В стоячем положении дети, чтобы сохранить равновесие, широко расставляют ноги и отклоняют туловище назад, так что замечается лордоз в поясничной части. При ходьбе бедра и туловище переваливаются из стороны в сторону (утиная походка). Подъем из лежачего положения возможен только с большим трудом; обыкновенно дети становятся при этом сперва на четвереньки и затем, упираясь руками в колени, постепенно передвигают руки по бедрам вверх и таким образом выпрямляются. Живот сильно выпячен, грудь уплощена.

При осмотре пораженные мышцы сильно увеличены в объеме, особенно на нижних конечностях. Мышцы наощупь то тверды и плотны, то мягки и как бы тестоваты, смотря по тому, преобладает ли в них склеротический или жировой процесс. Обыкновенно поражаются также мышцы туловища и верхних конечностей, иногда лица.
Псевдогипертрофия мышц, очень характерная для болезни, не всегда наблюдается. На верхних конечностях некоторые мышцы плеча представляют лишь простую атрофию, между тем как другие, особенно дельтовидная мышца, увеличены в объеме. Развивающиеся мышечные параличи могут быть полными, но они никогда не предшествуют атрофии и соответствуют всегда ее степени. Сухожильные рефлексы отсутствуют, когда мышцы парализованы. Фибриллярных подергиваний никогда не замечается, как это бывает при спинномозговых мышечных атрофиях. Кожная чувствительность нормальна. Электрическая возбудимость пораженных мышц более или менее понижена, но реакции перерождения никогда не бывает. Бульбарные явления всегда отсутствуют. В некоторых случаях наблюдается гипертрофия или атрофия щитовидной железы.
Мышцы бледны, желтоваты и нередко увеличены в объеме. При слабом увеличении видно под микроскопом, что мышечные волокна истончены, утратили на разрезе свою кругловатую форму и раздвинуты прослойками разросшейся соединительной ткани. При сильном увеличении замечается размножение ядер и скопление в окружности их саркоплазмы, маскирующей поперечную исчерченность.

Сильно разросшаяся соединительная ткань пропитана множеством жировых клеток. В менее пораженных мышцах наблюдаются, рядом с атрофированными мышечными волокнами, более или менее многочисленные гипертрофические (увеличенные в объеме) волокна. Сосуды не изменены; часто существующий периартериит находится в связи с разращением соединительной ткани. Центральная нервная система и периферические нервы не представляют никаких изменений.
Атрофия типа Лейден-Мобиуса
Эта форма развивается чаще у детей старшего возраста и клинически не отличается от формы, описанной Дюшеном, за исключением того, что пораженные мышцы не представляют псевдогипертрофии, а просто атрофированы. В некоторых случаях первоначально наблюдается ложная гипертрофия, которая в дальнейшем исчезает.
Миопатическая мышечная атрофия типа Ландузи-Дежерина (лице-лопаточно-плечевой тип)
При этой форме мышечная атрофия начинается с лица. Сперва атрофируется круговая мышца рта, вследствие чего ротовая щель расширяется, и нижняя губа отвисает; мало-помалу атрофия распространяется затем на другие лицевые мышцы, на круговую мышцу век, лобную мышцу и т. п. Лицо теряет выражение, больной не может полностью закрыть глаз, движения рта при свисте, смехе и разговоре затруднены. Язык и небная занавеска остаются непораженными. В дальнейшем атрофируются мышцы плечевого пояса, плеча, разгибатели спины, тазовые и бедренные мышцы. Течение очень хроническое (40 лет и более). Смерть может наступить от какой-либо присоединившейся инфекции.
Миопатическая мышечная атрофия типа Циммерлина
Атрофия начинается большей частью в юношеском возрасте и с мышц плечевого пояса, затем переходит на плечо. В остальном течение такое же, как при форме, описанной Дюшеном.
Мышечная атрофия типа Эрба (юношеская форма)
При этой форме бывают поражены некоторые мышцы плеча, крестца, таза, бедер и спины.

Сравнительно рано атрофируются мышцы плеча, сгибающие предплечье, затем большая грудная мышца, трапециевидная (позже всего ключичная ее часть), широкая спинная, большая зубчатая, ромбовидные и крестцово-поясничные мышцы. Напротив, дельтовидная, большая и малая круглые мышцы, над- и подостные, поднимающая угол лопатки, грудино-ключично-сосковая мышцы поражаются очень поздно, причем они представляют иногда ложную гипертрофию. В конце концов, атрофия распространяется постепенно на другие мышцы туловища и на мышцы бедер.
Диагноз
Болезнь Томсена, сопровождающаяся мышечной гипертрофией, может быть легко исключена на основании того, что при ней наблюдается тетаническая ригидность мышц в начале произвольных движений и получается миотоническая реакция.
Прогноз
Прогноз зависит от формы заболевания, стадии и лечения, в общем, он всего серьезен.
Двусторонняя атрофия мышц плечевого пояса наблюдается при миопатии, ревматоидном артрите, полимиозите (дерматомиозите). Односторонняя атрофия мышц плечевого пояса может возникнуть при плечелопаточном периартрите, анкилозе плечевого сустава.
Атрофия дельтовидной мышцы в относительно изолированном виде развивается при поражении подмышечного нерва (С5-6) по тем же причинам, что и поражение всего плечевого сплетения. Атрофия легко выявляется при осмотре плеча, и особенно при исследовании таких движений, как отведение плеча до горизонтальной плоскости, смещение его вперед, назад, страдает и супинация.
Атрофия поверхностных грудных мышц также обусловлена патологией плечевого сплетения (C5-D6), при этом существенно нарушается конфигурация передней грудной стенки, особенно у мужчин, западает подключичная область, более рельефной становится ключица, иногда начинают вырисовываться верхнегрудные ребра. Из-за слабости мышц больной не может крепко привести, прижать к туловищу плечо или энергично его отнять, ухудшается также тяга лопатки вперед и книзу, экскурсия грудной клетки. От атрофии следует отличать иногда встречающееся врожденное отсутствие большой грудной мышцы.
После осмотра плечевого пояса необходимо исследовать лопаточную область, имеющую с ним тесное функциональное взаимоотношение. Лопаточная область ограничена сверху уровнем VII шейного позвонка и акромиальиым отростком, снаружи — наружным краем дельтовидной мышцы и средней подмышечной линией, снизу — уровнем угла лопатки, внутри — внутренним краем лопатки.
Лопатка участвует в отведении верхней конечности, нижний угол лопатки при движении может подняться на 9-11 см вверх и сместиться до 19 см от позвоночника. Нарушение функции лопатки в первую очередь связано с нарушением функции передней зубчатой мышцы, которая тянет лопатку кнаружи и вперед, прижимает ее к ребрам, а также поражением большой и малой ромбовидных мышц. Эти мышцы при сокращении тянут лопатку к срединной плоскости и кверху. В приближении лопатки к позвоночнику участвуют средние и нижние волокна трапециевидной мышцы. Движение лопатки вверх осуществляется при участии мышцы, поднимающей лопатку. Иннервируется лопаточная область ветвями плечевого сплетения — надлопаточным и подлопаточным нервами.
Атрофия мышц туловища — глубоких, мышц спины, межреберных мышц, мышц живота наблюдается в результате перенесенного полиомиелита, при прогрессирующей мышечной атрофии. Поражение длинных мышц спины приводит к нарушению осанки, возникает гиперлордоз позвоночника в поясничном и шейном отделах (рис. 240).
Атрофия (гипотрофия) мышц верхних и нижних конечностей может быть ограниченной, касаться отдельной мышцы или группы мышц или охватывать всю конечность, что зависит от уровня поражения нервных стволов, артериальных сосудов, сухожилий и суставов.
Атрофия передней группы мышц плеча (двуглавая, клюво-плечевая, плечевая) может быть обусловлена поражением мышечно-кожного нерва (С6-С7), что приводит к нарушению сгибания руки в локтевом суставе и супинации предплечья, поднимания и приведения руки к срединной линии. Причинами могут быть сыпной тиф, пневмония, грипп, туберкулез, малярия. Неврит мышечно-кожного нерва возможен у официантов в силу профессиональных особенностей. Атрофия двуглавой мышцы развивается при длительном нарушении функции локтевого сустава.
Задняя группа мыши, плеча (трехглавая, локтевая) атрофируется при поражении лучевого нерва (С7-С8), что проявляется нарушением движения руки назад и разгибания предплечья, страдает приведение плеча к туловищу и разгибание предплечья в локтевом суставе.
Атрофия и слабость передней и латеральной группы мышц предплечья связано с поражением срединного нерва (C6-C7). Нарушается пронирование предплечья, сгибание в локтевом суставе. Задняя группа мышц предплечья страдает при поражении лучевого нерва (С6-С8), ухудшается или становится невозможной супинация предплечья, частично — разгибание руки в локтевом суставе. Атрофия мышц предплечья всегда приводит к нарушению определенных функций кисти — сгибание, разгибание, отведение, приведение, а также сгибание и разгибание пальцев.
И.А. Реуцкий, В.Ф. Маринин, А.В. Глотов
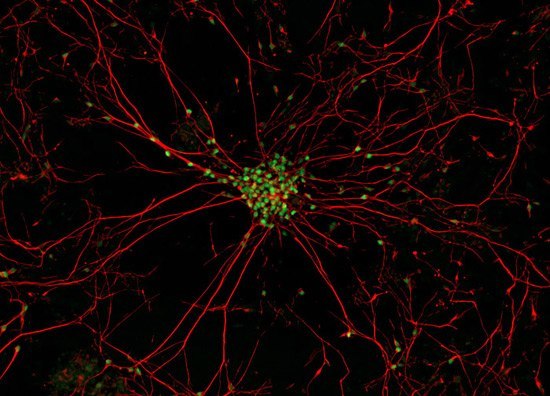
Настоящее фото мотонейрона (двигательного нейрона в передних рогах спинного мозга) - закупорка межсинаптических щелей (зелёные точки) - блокада передачи импульсов через медиаторы (вещества для передачи импульсов) в синапсы (места соединения) отростков нервных клеток.
В центре - тело мотонейрона.
Красные линии -длинные отростки мотонейрона - аксоны и короткие -дендриты.
Причина появления блокады передачи импульсов в межсинаптических щелях учёными мира не найдена. Предположительно - мутация гена,кодирующего фермент передачи этих импульсов через нейромедиаторы - супероксиддисмутазы. (СОД).
Предрасположенность к мутациям гена может иметь наследственный характер по аутосомно-рецессивному типу.
Эндемические (массовые вспышки) случаи этого заболевания зафиксированы у групп военных, живущих в островах на тихом океане. Чаще болеют мужчины от 40 до 60 лет.Следовательно не исключается инфекционная причина развития заболевания.
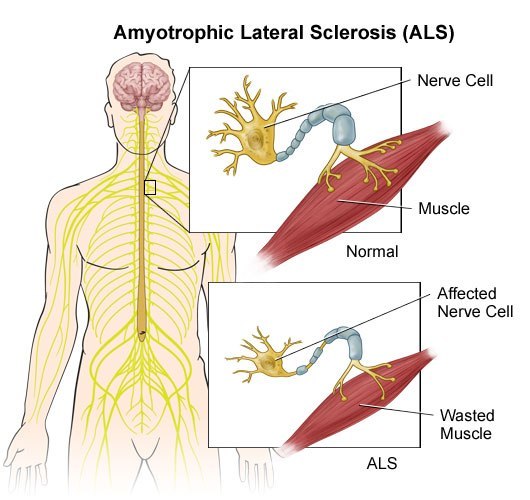
Боковой амиотрофический склероз. (БАС)..
Показано истончение нервных волокон в случае БАС и нарушение иннервации (передачи нервных импульсов) к мышцам. Как следствие - уменьшение работы мышцы и её последующая атрофия. (уменьшение размеров, обратное развитие.)
Блокада передачи нервных импульсов к мышцам (как к поперечно-полосатым которыми мы управляем сами своей волей так и к к гладким, работающим, независимо от нашего сознания, усилий и воли) пищеварительной и дыхательной системы ведёт к смерти из-за невозможности совершать эти жизненно важные моменты работы мускулатуры.
Две статьи из медицинских источников:
1) Теория аксостаза бокового амиотрофического склероза. Аксональная теория бокового амиотрофического склероза
Теория аксостаза основана на анализе патологических процессов, происходящих в аксональном транспорте мотонейронов [Chou S., 1992]. Наибольшими нейронами организма являются двигательные мотонейроны передних рогов спинного мозга и пирамвды Беца. Они должны поддерживать интеграцию дендритов, часто протяженностью более 1 см, и аксон, достигающий 100 см. В аксоне имеются непрерывные потоки, через которые клеточное тело направляет структурные и функциональные белки на периферию и получает обратные сигналы. Ортоградный транспорт бывает 2 видов: а) быстрый — 400 мм в день, идущий в обоих направлениях и транспортирующий связанные с мембраной белки и гликопротеиды, б) медленный — несколько миллиметров в день, транспортирующий сети микрофиламентов, микротрубочек, нейрофиламентов, как компонент "а" (0,1—2 мм в день), а также большой комплекс растворимых белков, как компонент "б" (2—4 мм в день). Ретроградный аксональный транспорт несет эндогенные (аминокислоты, фактор роста нервов) и экзогенные (токсин столбняка, вирус полиомиелита, простого герпеса, бешенства, лектин пероксидазы хрена и др.) субстанции от терминальных аксонов к клеточному телу со скоростью свыше 75 мм в день. Морфологические исследования аксонального транспорта в биоптатах двигательных веточек периферических нервов больных боковым амиотрофическим склерозом выявили уменьшение скорости ретроградного аксонального транспорта и, следовательно, связи терминального аксона с перикарионом [Bieuer A. et al., 1987]. В межреберных нервах больных АБС еще до развития признаков нейрональной дегенерации появляются изменения белков микротрубочек [Binet S. et al., 1988].
Улыраструктурные исследования проксимального аксона и аксонального бугорка мотонейронов переднего рога спинного мозга больных, умерших от бокового амиотрофического склероза [Sasaki S. et al., 1996], показали нарушение быстрого аксонального транспорта. Гладкий эндо-плазматический ретикулум теряет структуру: происходит скопление митохондрий, лизосом, Леви-подобных телец, эозино-фильных и гиалиновых включений, липофусциновых гранул, особенно в аксональном бугорке. Присутствие этих необычных структур является отражением дисфункции аксонального транспорта. Применительно к возможной этиологии АБС еще ранее выдвинута концепция "аксостаза" [Chou S., 1992]. Ней-ротоксические факторы путем ретроградного транспорта избирательно поражают нейрон, создавая феномен "суицидцального транспорта". Ухудшение медленного транспорта в аксоне сопровождается скоплением нейрофиламентов, набуханием проксимального аксона и последующей дистальнои аксональной атрофией, а также вторичной демиелинизацией, характерной для центральной дистальнои аксонопатии или "ретроградного умирания" — "dying back". Определенную значимость в развитии ранних морфологических изменений мотонейронов при АБС имеет теория аутоиммунитета [Smith R. et al., 1996], основанная на появлении антител к зарядам входа кальциевых каналов. Пассивный перенос фракций, содержащих иммуноглобулин, мышам вызывает изменения нервно-мышечных соединений, сходные с таковыми при спорадическом АБС. У животных эти изменения отражают расстройства внутриклеточного Са2+ гомеостаза, и раннее повреждение пластинчатого комплекса в мотонейронах в форме набухания и фрагментации. Иммуноглобулины от больных спорадическим боковым амиотрофическим склерозом вызывают зависимый от Са2+ апоптоз клеток вследствие оксидативных повреждений. Апоптоз, обусловленный иммуноглобулином от указанных больных, регулируется присутствием связанных белков, которые могут модулировать избирательную ранимость нейронов при спорадическом АБС.
2) Боковой амиотрофический склероз
Несмотря на более чем 100-летнее изучение, боковой амиотрофический склероз (БАС) остается фатальным заболеванием центральной нервной системы. Заболевание характеризуется неуклонно прогрессирующим течением с избирательным поражением верхнего и нижнего мотонейронов, что приводит к развитию амиотрофий, параличей и спастичности. До настоящего времени вопросы этиологии и патогенеза остаются невыясненными, в связи с чем не разработаны специфические методы диагностики и лечения этого заболевания. Рядом авторов отмечено повышение частоты встречаемости заболевания среди лиц молодого возраста (до 40 лет).
МКБ-10 G12.2 Болезнь двигательного неврона
ЭПИДЕМИОЛОГИЯ
Боковой амиотрофический склероз дебютирует в возрасте 40 – 60 лет. Средний возраст начала заболевания 56 лет. БАС - болезнь взрослых, и не наблюдается у лиц моложе 16 лет. Несколько чаще заболевают мужчины (отношение мужчины-женщины 1,6-3.0: 1).
БАС является спорадическим заболеванием и встречается с частотой 1,5 – 5 случая на 100 000 населения.
В 90% случаев БАС носит спорадический, а в 10% - семейный или наследственный характер как с аутосомно-доминантным (преимущественно), так и с аутосомно-рецессивным типами наследования. Клинические и патоморфологические характеристики семейного и спорадического БАС практически идентичны.
В настоящее время возраст является основным фактором риска при БАС, что подтверждается нарастанием заболеваемости после 55 лет, и в этой возрастной группе уже не наблюдается различий между мужчинами и женщинами. Несмотря на достоверную связь БАС с возрастом, старение является только одним из предрасполагающих факторов развития патологического процесса. Вариабельность заболевания как в различных возрастных группах, так и среди лиц одного возраста предполагает существование определённых факторов риска: дефицит, или наоборот, наличие определённых нейропротективных факторов, к которым в настоящее время относят: нейростероиды или половые гормоны; нейротрофические факторы; антиоксиданты.
Некоторые исследователи отмечают особо благоприятное течение заболевания у молодых женщин, что подтверждает несомненную роль половых гормонов, в особенности эстрадиола и прогестина, в патогенезе бокового амиотрофического склероза. Подтверждением этому являются: большая частота встречаемости БАС у мужчин до 55 лет (при этом у них отмечается более раннее начало и быстрое прогрессирование заболевания по сравнению с женщинами); с наступлением менопаузы женщины болеют также часто, как и мужчины; единичные случаи заболевания боковым амиотрофическим склерозом во время беременности. К настоящему времени существуют единичные работы по изучению гормонального статуса больных с боковым амиотрофическим склерозом, и ни одной, посвящённой определению концентраций гормонов у молодых пациентов.
Этиология заболевания не ясна. Обсуждается роль вирусов, иммунологических и метаболических нарушений.
В развитии семейной формы БАС показана роль мутации в гене супероксиддисмутазы-1 (Cu/Zn-супероксиддисмутазу, SOD1), 21q22-1 хромосома, выявлен также БАС, связанный с 2q33-q35 хромосомой.
Синдромы, клинически не отличимые от классического БАС, могут возникать в результате:
•опухоли большого затылочного отверстия
•спондилез шейного отдела позвоночника
•артериовенозная аномалия спинного мозга
•бактериальные - столбняк, болезнь Лайма
•вирусные - полиомиелит, опоясывающий лишай
Интоксикации, физические агенты:
•токсины - свинец, алюминий, другие металлы.

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Большинство из обсуждаемых здесь заболеваний приводят к двусторонней проксимальной слабости и атрофии симметричного характера (за исключением проксимальной диабетической полинеиропатии, невралгической амиотрофии и, частично, боковой амиотрофический склероз) на руках и на ногах. Здесь не обсуждаются синдромы поражения плечевого и пояснично-крестцового сплетений (плексопатии), которые чаще встречаются с одной стороны.
Проксимальная мышечная слабость может наблюдаться преимущественно в руках, преимущественно в ногах или развиваться генерализованно (и в руках, и в ногах).
Преимущественно в руках проксимальная слабость мышц может быть иногда проявлением бокового амиотрофического синдрома; некоторых форм миопатий (в том числе воспалительных); ранних стадий синдрома Гийена-Барре; синдрома Персонейджа-Тернера (чаще одностороннего); полинейропатии, связанной с гипогликемией; амилоидной полинеиропатии и некоторых других формах полинеиропатии.
Проксимальная слабость мышц преимущественно в ногах может быть обусловлена почти теми же самыми заболеваниями; некоторыми формами миопатий; полинейропатии (диабетическая, некоторые токсические и метаболические формы), полимиозитом, дерматомиозитом, некоторыми формами прогрессирующей спинальной амиотрофии. Некоторые из перечисленных заболеваний могут одновременно или последовательно вызывать проксимальную слабость как в руках, так и в ногах.

[1], [2], [3], [4], [5], [6], [7], [8], [9]
Основные причины мышечной проксимальной слабости:
- Миопатия (несколько вариантов).
- Полимиозит (дерматомиозит).
- Проксимальная диабетическая полинейропатия.
- Невралгическая амиотрофия.
- Миелит.
- Синдром Гийена-Барре и другие полинеиропатии.
- Боковой амиотрофический склероз.
- Проксимальные формы прогрессирующей спинальной амиотофии.
- Паранеопластическая болезнь моторного нейрона.
При постепенном развитии двусторонней проксимальной мышечной слабости в проксимальных отделах конечностей, прежде всего, следует думать о миопатии. Начальная стадия заболевания характеризуется наличием мышечной слабости, степень которой существенно превосходит незначительно выраженные атрофии соответствующих мышц. Фасцикуляции отсутствуют, глубокие рефлексы с конечностей сохранены или немного снижены. В чувствительной сфере никаких изменений нет. При физической нагрузке пациент может испытывать боль, что указывает на довольно распространенное вовлечение соответствующих мышечных групп в патологический процесс и свидетельствует о нарушении функционирования нормального механизма поочередного включения работающей и отдыхающей порции мышцы (мышц).
Миопатия - это еще не диагноз; данный термин лишь указывает на мышечный уровень поражения. Далеко не все миопатии имеют дегенеративный характер. Уточнение характера миопатии позволяет выработать соответствующую тактику лечения. Некоторые миопатии являются проявлением потенциально курабельных заболеваний, например - метаболических расстройств или аутоиммунных заболеваний.
Довольно ценную информацию о возможной причине миопатии могут дать лабораторные исследования. Наиболее информативно изучение биоптатов мышц. Помимо исследования миобиоптата методами световой или электронной микроскопии абсолютно необходимо применение современных ферментативных гистохимических и иммунохимических исследований.
Особняком от основной группы полимиозитов стоят воспалительные процессы в мышцах, вызванные конкретными микроорганизмами. Примером может быть миозит вирусной природы, характеризующийся острым началом с выраженной боли и очень высокими показателями СОЭ. Выраженные болевые проявления также характерны для ограниченного миозита при саркоидозе и трихинеллезе. Это свойственно и ревматической полимиалгии (polymyalgia rheumatica) - заболеванию мышц, возникающему в зрелом и пожилом возрасте и протекающему с выраженным болевым синдромом. Истинная мышечная слабость, как правило, отсутствует или выражена минимально - движения затруднены из-за интенсивной боли, особенно - в мышцах плечевого и тазового пояса. ЭМГ и биопсия не выявляют признаков повреждения мышечных волокон. СОЭ значительно повышена (50-100 мм в час), лабораторные показатели указывают на подострый воспалительный процесс, КФК чаще нормальна. Возможна слабо выраженная анемия. Характерен быстрый эффект кортикостероидов. У некоторых пациентов в последующем возникает артериит краниальной локализации (височный артериит).
Проксимальная мышечная слабость может быть проявлением патологии периферической нервной системы, чаще всего - диабетической нейропатии. Такой клинический вариант диабетической полинейропатии с вовлечением проксимальных мышечных групп гораздо менее известен врачам в отличие от хорошо известной формы диабетической полинейропатии, при которой имеется двусторонний симметричный дистальный сенсомоторный дефект. У части пациентов зрелого возраста, страдающих диабетом, возникает проксимальная слабость в конечностях, как правило - асимметричная, часто присутствует боль, но наиболее очевидный двигательный дефект - слабость и проксимальная атрофия. Затруднены подъем и спускание с лестницы, вставание из положения сидя, переход в сидячее положение из положения лежа на спине. Ахилловы рефлексы могут оставаться сохранными, но коленные рефлексы, как правило, отсутствуют; четырехглавая мышца бедра болезненна при пальпации, паретична и гипотрофична. Выявляется слабость в m. ileopsoas. (Близкую картину асимметричной проксимальной слабости и атрофии дают такие заболевания, как карциноматозная или лимфоматозная радикулопатия).
Для развития проксимальной диабетической полинейропатии (равно как для развития всех других форм диабетической нейропатии) совсем не обязательно наличие тяжелых метаболических нарушений: иногда они могут выявляться впервые при проведении глюкозо-толерантного теста (латентный диабет).
Асимметричную проксимальную диабетическую полинейропатию в нижних конечностях следует отличать от одностороннего поражения поясничного сплетения - заболевания, аналогичного хорошо известной невралгической амиотрофии мышц плечевого пояса. Клинические наблюдения последних 10 лет показали, что аналогичный патологический процесс может поражать и поясничное сплетение. Клиническая картина представлена симптомами острого одностороннего поражения бедренного нерва с развитием паралича иннервируемых им мышц. При тщательном исследовании, включая ЭМГ и исследование скорости проведения по нервам, можно также обнаружить легкое вовлечение соседних нервов, например - запирательного нерва, что проявляется в виде слабости приводящих мышц бедра. Заболевание имеет доброкачественный характер, выздоровление наступает через несколько недель или месяцев.
Крайне важно убедиться в отсутствии у пациента двух других возможных заболеваний, требующих специфического диагностического подхода и лечения. Первое - это повреждение третьего или четвертого поясничных спинно-мозговых корешков: в этом случае не нарушается потоотделение на передней поверхности верхней части бедра, так как вегетативные волокна покидают спинной мозг в составе корешков не ниже второго поясничного.
Потоотделение нарушается при злокачественных новообразованиях в тазу, воздействующих на поясничное сплетение, через которое проходят вегетативные волокна. Другая причина сдавления поясничного сплетения, которую следует иметь в виду, это - спонтанная забрюшинная гематома у пациентов, получающих антикоагулянты. В такой ситуации у пациента возникает боль вследствие начального сдавления гематомой бедренного нерва; для снятия боли пациент принимает анальгетики, анальгетики усиливают эффект антикоагулянтов, что ведет к дальнейшему увеличению объема гематомы и давления на бедренный нерв с последующим развитием паралича.
Случаи миелита с развитием проксимальных парезов стали редкими с тех пор, как из клинической практики практически исчез полиомиелит. Другие вирусные инфекции, вызванные, например, вирусом Коксаки типа А, могут имитировать полиомиелитический неврологический синдром, приводя к развитию асимметричного проксимального пареза с отсутствием рефлексов при сохранной чувствительности. В ликворе выявляют повышенный цитоз, легкое повышение уровня белка и относительно низкий уровень лактата.
Описанный выше миелит следует дифференцировать с синдромом Гийена-Барре, что в первые дни заболевания представляется весьма трудной задачей. Неврологические проявления очень сходные - даже поражение лицевого нерва может наблюдаться при обоих заболеваниях. Скорости проведения по нервам в первые дни могут оставаться нормальными, то же относится и к уровню белка в ликворе. В пользу миелита свидетельствует плеоцитоз, хотя он обнаруживается и при синдроме Гийена-Барре, в частности - при синдроме Гийена-Барре вирусной природы (напр., вызванном вирусом Эпштейна-Барр). Вовлечение вегетативной нервной системы является важным диагностическим критерием, свидетельствующим в пользу синдрома Гийена-Барре, если доказана ареактивность частоты сердечных сокращений на стимуляцию блуждающего нерва или выявлены другие симптомы периферической вегетативной недостаточности. Дисфункция мочевого пузыря наблюдается при обоих патологических состояниях, то же относится и к параличу дыхательных мышц. Иногда только наблюдение за течением заболевания с повторной оценкой неврологического статуса и скоростей проведения по нервам позволяет правильно поставить диагноз. Для некоторых других форм полинейропатии также характерна преимущественно проксимальная акцентуация процесса (полинейропатия при лечении винкристином, при контакте кожи с ртутью, полинейропатия при гигантоклеточном артериите). ХВДП иногда проявляется подобной картиной.
Дебют бокового амиоторофичечского склероза с проксимальных отделов руки - явление не частое, но вполне возможное. Асимметричная амиотофия (в начале заболевания) с гиперефлексией (и фасцикуляциями) - характерный клинический маркёр бокового амиотрофического склероза. ЭМГ выявляет переднероговую заинтересованность даже в клиничнески сохранных мышцах. Заболевание неуклонно прогрессирует.
[10], [11], [12], [13], [14], [15], [16], [17]
Некоторые формы прогрессирующей спинальной амиотрофии (Вердниг-Гофмана амиотрофия, Кугельберга-Веландер амиотрофия) относятся к проксимальным спинальным амиторофиям наследственного характера. Фасцикуляции имеются не всегда. Сфинктерные функции сохранны. Для диагноза важнейшее значение имеет ЭМГ. Проводниковые системы спинного мозга, как правило, не вовлекаются.
Паранеопластическая болезнь моторного нейрона (поражение спинного мозга) может иногда имитировать прогрессирующую спинальную амиотрофию.
Как распознается проксимальная мышечная слабость?
Общий и биохимический анализ крови; анализ мочи; ЭМГ; биопсия мышц; исследование уровня КФК в крови; определение скорости проведения возбуждения по нерву; исследование ликвора; консультация терапевта; при необходимости - онкопоиск и другие (по показаниям) исследования.
Читайте также:
