
Мышцы ног генетическое заболевание
Спинальная мышечная атрофия относится к генетически обусловленным заболеваниям и характеризуется прогрессирующей атрофией мышц в результате нарушения периферической иннервации. При этом происходит блокада импульсов спинного мозга, идущих к мышечным волокнам, что вызывает прекращение их нормального развития и функционирования. Чаще поражаются мышцы туловища и проксимальных отделов конечностей. Болезнь развивается у новорожденных детей, но иногда поражает людей молодого возраста. 
Причины развития болезни
Развитие патологии связывают с мутацией генов, которая приводит к нарушению синтеза специфического белка, участвующего в жизнедеятельности двигательных нейронов спинного мозга. Они представляют собой скопление клеток, которые локализуются в передних спинномозговых отделах и образуют двигательные корешки. Главной функцией нейронов считается регулирование сократительной активности, кровообращения, обменных процессов мышечных волокон.
При нарушении синтеза белка мотонейроны постепенно разрушаются как в правой, так и левой половине спинного мозга. Симметричная спинная мышечная атрофия относится к важному диагностическому признаку.
В результате к мышцам не поступают сигналы из центральной нервной системы, они теряют способность к сокращению. Это негативно влияет на двигательную активность и приобретение ребенком навыков самообслуживания. При этом чувствительная иннервация сохраняется в полном объеме.
Заболевание передается по аутосомно-рецессивному типу, поэтому встречается в популяции населения крайне редко. Для того чтобы патология начала развиваться в организме, необходимо носительство дефектного гена обоими родителями. При этом чаще всего у них нет проявлений заболевания, а обоюдное носительство не всегда дает появление недуга у совместного ребенка. Доказано, что каждый 40 человек может содержать в своем генетическом материале мутации определенного вида хромосом, обуславливающие носительство дефектного гена.
Проявления заболевания
Выделяют несколько форм развития патологии, которые отличаются возрастом возникновения первых симптомов, тяжестью течения, продолжительностью жизни.
Чем позже развивается патология, тем лучше прогноз для жизни и поддержания навыков самообслуживания.
В любом случае больные становятся инвалидами, требующими дополнительный уход и специальных средств передвижения (коляски, ходунки). Часто пациенты прикованы к кровати, их состояние осложняется застойными процессами в легких, которые могут привести к летальному исходу.

Спинальная мышечная атрофия тип 1 может быть заподозрена еще во внутриутробном периоде при слабом шевелении ребенка во второй половине беременности. На протяжении полугода после рождения отмечают вялость, низкую активность новорожденного. При обследовании не обнаруживают сухожильные рефлексы (ахиллов, коленный).
Мышцы верхних и нижних конечностей, туловища и головы не развиваются. Слабость и атрофия мышечных волокон приводит к неспособности детей полноценно сидеть, ползать, вставать на ноги. По мере роста скелета возникает деформация костей. Например, при попытке новорожденного сидеть позвоночник не поддерживается мышечным каркасом спины, что приводит к развитию кифоза.
Нередко выявляют непроизвольное подергивание мышц, дрожание пальцев вытянутых рук. Мышечная атрофия может визуально сглаживаться чрезмерным развитием подкожно-жировой клетчатки. Из-за недоразвития межреберных мышц грудная клетка приобретает уплощенную форму. Страдает функция глотания, сосания, дыхания, в результате чего развиваются тяжелые осложнения: аспирационная и застойная пневмония, истощение, дыхательная недостаточность.
Развитие умственной сферы проходит удовлетворительно, так как отделы головного мозга не подвергаются патологическим изменениям.
Эта форма заболевания считается неблагоприятной для жизни, такие дети обычно погибают в течение первых лет после рождения.
Атрофия спинного мозга в случае 2 типа болезни проявляется в конце первого года жизни, обычно в 7−18 месяцев ребенка. Признаки патологии выражены не резко и развиваются постепенно. По мере взросления новорожденных отмечают слабую двигательную активность. Дети предпочитают лежать, неохотно перемещаются в пространстве, значительно отстают в физическом развитии от своих сверстников.
Мышцы тела и конечностей подвержены непроизвольным сокращениям, отмечают подергивание языка. Дети способны сидеть, вставать на ноги, иногда медленно передвигаться с посторонней помощью. Прием пищи и дыхание обычно не затруднено, сухожильные рефлексы развиты слабо. Продолжительность жизни таких больных снижают частые респираторные инфекции и тяжелые застойные воспаления легких в результате вялой двигательной активности.
Первые симптомы недуга появляются в конце второго года жизни ребенка, иногда развитие мышечной атрофии начинается в период полового созревания. Заболевание характеризуется медленным прогрессированием. Сначала атрофируются верхние мышечные группы ног, затем патологический процесс поражает руки, туловище, шею. 
Больные, страдающие 3 типом болезни, способны передвигаться: вставать, ходить, подниматься по лестнице. Характерным признаком заболевания считается гипертрофия ягодичных и икроножных мышц. Однако по мере нарастания дистрофических изменений в передних отделах спинного мозга сухожильные рефлексы угасают, что свидетельствует о необратимых изменениях мышечной ткани.
Со временем двигательная активность снижается, что заставляет больных пользоваться инвалидными колясками. Длительно сохраняется способность к самообслуживанию, тазовые функции развиты хорошо (мочеиспускание, дефекация). Чувствительная иннервация при заболевании не страдает.
Терапевтическая тактика
Лечение спинальной мышечной атрофии имеет поддерживающий характер и проводится в течение всей жизни больного.
Выздоровления достичь невозможно, а лишь замедлить развитие патологии в нервной и мышечной ткани.
Терапевтическая тактика включает комплекс мер, направленных на сохранении двигательной активности.
- Лекарственные средства, нормализующие проведение нервного импульса — прозерин, нивалин.
- Ноотропные препараты, улучшающие кровообращение нервной ткани — пирацетам, ноотропил.
- Средства, стимулирующие кровоток и обменные реакции в мышечной ткани — актовегин, оротат калия.
- Витаминные препараты, обладающие антиоксидантным действием — токоферол, витамины группы В, глутаминовая кислота.
- Физиопроцедуры, усиливающие кровообращения пораженных конечностей — парафин, УВЧ, электрофорез с прозерином.
- Массаж — стимулирует мышечную активность, способствует выведению продуктов обмена.
- Лечебная гимнастика — атрофия мышц спины, рук, ног после занятий замедляется.
- Ортопедическая помощь — использование специальных приспособлений, поддерживающих двигательную активность грудной клетки, верхних и нижних конечностей.
В терминальных стадиях заболевания появляются дыхательные расстройства, которые требуют перевода пациентов на искусственную вентиляцию легких.
Спинальная мышечная атрофия считается тяжелым генетическим заболеванием с постоянным прогрессированием патологического процесса и неблагоприятным прогнозом.
В последние годы ученые всего мира ведут научные разработки по синтезированию лекарственного препарата, способного замещать недостающий нейронный белок. Эта единственная надежда больных на выздоровление и значительное улучшение качества жизни.
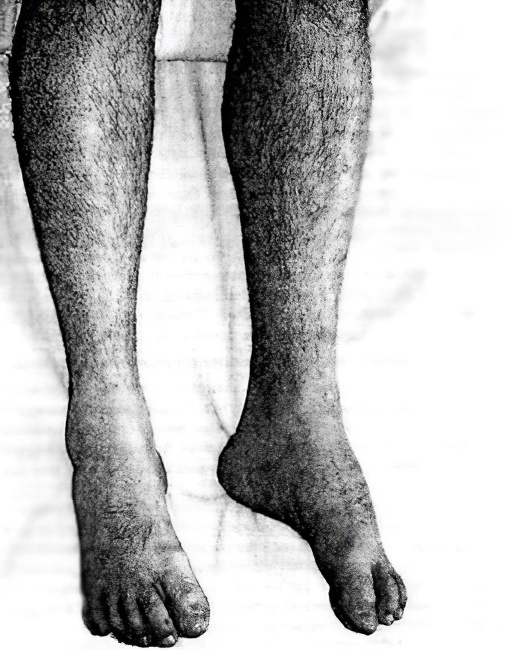
Вследствие патологических изменений в организме человека скелетная мышца начинает истончаться, деформироваться, затем происходит её замещение соединительной тканью, неспособной к сокращению, то есть происходит атрофия мышц. Как результат снижается двигательная способность пораженной мышцы, а при значительном её перерождении происходит полный паралич, больной теряет способность самостоятельно передвигаться.
Причин для развития атрофии мышц нижних конечностей может быть несколько:
- снижение метаболизма и старение организма с возрастом;
- как результат заболеваний эндокринной системы и гормонального сбоя в организме;
- хронические заболевания пищеварительного тракта, соединительной ткани;
- нарушение регуляции мышечного тонуса при поражении периферических нервов, полиневритах, как проявление осложнений некоторых инфекционных и паразитарных заболеваний, хронических отравлениях;
- плохая наследственность – врожденная ферментопатия или генетические нарушения;
- неполноценное, недостаточное питание;
- как посттравматические осложнения или при постоянной физической нагрузке.
Заболевания, связанные с атрофией мышц, как правило, относятся к редким врождённым генетическим заболеваниям, которые проявляться начинают уже в детстве.
В самом начале заболевания характерным симптомом является быстрая утомляемость в ногах, мышечная слабость при длительной физической нагрузке. Заметно увеличиваются икроножные мышцы. Атрофия обычно начинается с проксимальных (ближайших к телу) групп мышц нижних конечностей. Проявляется это в ограничении двигательной функции ног – больному тяжело подниматься по лестнице и вставать из горизонтального положения. Со временем изменяется походка.
Атрофия мышц развивается медленно и длится годами. Болезнь может распространяться как на одну, так и на обе стороны; процесс может быть как симметричным, так и ассиметричным. Все проявления зависят от причин и формы заболевания, возраста и состояния организма пациента. Клинические проявления заключаются в нарастающей слабости в нижних конечностях, появляется дрожание. Больные испытывают неприятные ощущения, чувство ползанья мурашек под кожей.
Самым характерным признаком развивающейся мышечной атрофии является уменьшение в объёме пораженной мышцы, что замечается даже самим больным на ранней стадии заболевания. Все труднее становится передвигаться без посторонней помощи, особенно тяжело подниматься и спускаться по лестнице. Заболевание протекает хронически, отмечаются периоды рецидивов (с сильными болями в пораженной мышце) и ремиссий с незначительным угасанием симптоматики.
Для первичной формы атрофии мышц характерно поражение самой мышцы, её двигательных нейронов, обусловленное неблагоприятной наследственностью или рядом других причин – травмами, ушибами, физическим перенапряжением. Больной очень быстро утомляется, мускулатура теряет тонус, характерны непроизвольные подергивания конечностей.
Вторичное поражение мышечной ткани нижних конечностей называется невральной амиотрофией, наиболее часто является последствием травм или перенесенных инфекционных заболеваний, как следствие генетической патологии. При этом страдают мышцы голеней и стоп, происходит их деформация. Стопа как будто бы висит, и чтобы не цепляться ею за пол человек начинает высоко поднимать колени при ходьбе. По мере прогрессирования и распространения процесса, атрофия мышц с ног переходит на кисти рук и предплечья.
Как правило, дети с данным врожденным генетическим заболеванием не доживают до 14 лет.
Патология сопровождается также изменениями со стороны сердечной мышцы, поражается головной мозг, ребенок отстает в развитии. Слабость дыхательных мышц становится причиной плохой вентиляции легких, что способствует развитию пневмонии. Течение пневмонии осложняется слабостью сердечной мышцы, что является самой распространенной причиной смерти пациентов. Для формы Дюшенна характерно плейотропное влияние патологического гена.
В середине ХХ века Беккером был описан доброкачественный вариант миопатии, сцепленной с полом, эта форма заболевания носит его имя. Первые симптомы патологии появляются после 20 лет. На начальном этапе заметна псевдогипертрофия икроножных мышц. Атрофия мышц ног развивается медленно, постепенно охватывая мышцы тазового пояса и бедер. Интеллект при этой форме сохраняется. Эти разновидности заболевания характеризуются повреждениями различных генов, располагающихся в двух локусах половой Х-хромосомы, являясь генокопиями. В одной семье сразу две формы заболевания не встречаются.
Для того, чтобы диагностировать атрофию мышц необходимо собрать тщательный анамнез, в том числе и узнать о наследственных и хронических заболеваниях. Назначается развернутый анализ крови с обязательным определением СОЭ, глюкозы, печеночных проб. Обязательна электромиография и иногда биопсия нервных клеток, а также исследование нервной проводимости. При наличии в анамнезе хронических заболеваний или перенесенных инфекционных, по показаниям проводится дополнительное обследование.
При выборе лечения основное внимание уделяют причинам, из-за которых развилось заболевание. Учитывается возраст пациента, распространенность и тяжесть патологического процесса. Медикаментозное лечение, проводимое курсами, способно приостановить процесс и даже приводит к некоторым улучшениям. Немаловажную роль играет назначение физиотерапевтического лечения, лечебного массажа, электролечения, лечебной гимнастики. Также при лечении атрофии мышц нередко практикуют переливание крови. Соблюдение всех рекомендаций позволяет больным вести практически нормальный образ жизни на протяжении длительного времени.

Эксперт-редактор: Мочалов Павел Александрович | д. м. н. терапевт
Образование: Московский медицинский институт им. И. М. Сеченова, специальность - "Лечебное дело" в 1991 году, в 1993 году "Профессиональные болезни", в 1996 году "Терапия".
24.1. Нервно-мышечные заболевания
Наследственные нервно-мышечные заболевания – большая гетерогенная группа болезней, в основе которых лежит генетически детерминированное поражение нервно-мышечного аппарата. Заболевания характеризуются мышечной слабостью, мышечными атрофиями, нарушениями статических и локомоторных функций.
При постановке диагноза учитываются возраст проявления первых клинических симптомов заболевания, локализация атрофии и характер распространения миодистрофического процесса (восходящий, нисходящий, наличие или отсутствие псевдогипертрофий, фасцикуляций, расстройств чувствительности, пароксизмов мышечной слабости), а также темп течения.
Прогрессирующие мышечные дистрофии – наиболее обширная группа. В зависимости от характера первичных изменений условно различают первичные (миопатии) и вторичные формы прогрессирующих мышечных дистрофий (денервационные амиотрофии – спинальные и невральные).
Наследственные пароксизмальные миоплегии – группа нервно-мышечных заболеваний, характеризующихся внезапными приступами мышечной слабости и плегиями. Наиболее распространенными из наследственных пароксизмальных миоплегии являются гипо-, гипер– и нормокалиемическая формы. Патогенез неясен. Предполагается генетически детерминированный дефект мембраны сарколеммы, нарушающий проницаемость для ионов натрия и калия,
Гипокалиемическая форма пароксизмальной миоплегии (болезнь Вестфаля). Заболевание описано Вестфалем в 1895 г. Наследуется по аутосомно-доминантному типу.
Клинические проявления. Болезнь проявляется в возрасте 6—15 лет. Пароксизмы характеризуются внезапным в ночные или утренние часы развитием мышечной слабости, обездвиженности, снижением мышечного тонуса, сухожильных рефлексов, вегетативными расстройствами – лабильностью пульса, артериального давления, гипергидрозом. Приступы бывают парциальными, охватывающими небольшую группу мышц, и генерализованными. Во время приступа возникают нарушения сердечно-сосудистой деятельности: систолический шум, изменения ЭКГ. Сознание всегда сохранено. Средняя продолжительность приступа – несколько часов, крайне редко пароксизмы держатся несколько суток. Содержание калия в крови во время приступа менее 2 ммоль/л и ниже. Частота приступов вариабельна. Они провоцируются перееданием пищи, богатой углеводами, охлаждением, физическими нагрузками.
Лечение. Диета, богатая калием (чернослив, курага, картофель, изюм). Для купирования приступа назначают 10% раствор хлорида калия внутрь (по 1 столовой ложке каждый час) или 0,5% раствор в изотоническом растворе хлорида натрия внутривенно (2—2,5 г на 500 мл раствора в течение часа). Целесообразно применять также панангин внутривенно капельно.
Гиперкалиемическая форма пароксизмальной миоплегин (болезнь Гамсторп). Заболевание описано И.Гамсторп в 1956 г. Наследуется по аутссомно-доминантному типу.
Клинические проявления. Болезнь проявляется в возрасте 1—5 лет. Симптоматика сходна с пароксизмами при гипокалиемической форме и характеризуется внезапным развитием мышечной слабости, плегиями, снижением мышечного тонуса, сухожильных рефлексов, вегетативными расстройствами. В отличие от гипокалиемического гиперкалиемический паралич развивается обычно днем, сопровождается выраженными парестезиями, сочетается со слабостью мышц лица, артикуляционного аппарата, имеет меньшую продолжительность (30—40 мин). Во время приступа содержание калия в крови повышается до 6—7 ммоль/л. Частота приступов вариабельна: от ежедневных до нескольких раз в месяц. В межприступные периоды неврологическая симптоматика отсутствует. Провоцирующими факторами являются голодание, физические нагрузки, вызывающие утомление.
Лечение. Диета с повышенным содержанием углеводов, поваренной соли, ограниченным количеством калия. Вводят 40 мл 40% раствора глюкозы внутривенно вместе с инсулином подкожно; 20 мл 10% раствора хлорида кальция внутривенно.
Нормокалиемический (периодический) паралич. Наследуется по аутосомно-доминантному типу.
Клинические проявления. Болезнь проявляется до 10-летнего возраста. Особенностью ее является сравнительно медленно (в течение нескольких суток) пароксизмально нарастающая умеренная слабость в мышцах туловища, конечностей и в жевательной мускулатуре, а также медленный (1—2 нед) регресс симптоматики. Провоцирующими факторами являются продолжительный сон, длительное пребывание в одной позе, переохлаждение.
Лечение. Диета, богатая поваренной солью. Назначают ацетазоламид (диакарб).
Течение. Все формы пароксизмальных миоплегий медленно прогрессируют. Прогноз при своевременно поставленном диагнозе, проведении экстренных мероприятий и дифференцированной медикаментозной терапии благоприятный.
Диагностика и дифференциальный диагноз. Диагноз строится на основании генеалогического анализа, особенностей клинической картины, с учетом возраста, в котором начинается заболевание, времени возникновения пароксизма (ночью, утром, днем, в неопределенное время), степени выраженности мышечной слабости, частоты и длительности приступа, провоцирующих факторов, данных лабораторного биохимического исследования (содержание биоэлектрической активности мышц).
Дифференцировать заболевание следует от миоплегий, развивающихся в результате первичных эндокринных заболеваний, – тиреотоксикоза, болезни Конна (первичный гиперальдостеронизм), болезни Аддисона и др.
Лечение. Показана диета, богатая поваренной солью. Назначают диакарб.
24.2. Пирамидные и экстрапирамидные дегенерации
Хроническое прогрессирующее заболевание нервной системы, клинически проявляющееся изменениями мышечного тонуса и непроизвольными тоническими сокращениями мышц туловища и конечностей.
Этиология и патогенез. Различают идиопатическую (семейную) торсионную и симптоматическую дистонию. Тип наследования при идиопатической торсионной дистонии как аутосомно-доминантный, так и аутосомно-рецессивный. Симптоматическая торсионная дистония встречается при гепатоцеребральной дистрофии, хорее Гентингтона, опухолях мозга, эпидемическом энцефалите, детском церебральном параличе. Имеются указания, что в патогенезе наследственной торсионной дистонии имеет значение нарушение допаминового обмена. При обследовании у этих больных обнаруживается повышение содержания допамин-?-гидроксилазы в сыворотке крови.
Патоморфология. Дистрофические изменения обнаруживаются преимущественно в мелких нейронах в области скорлупы чечевицеобразного ядра, реже – в других базальных ганглиях.
Клинические проявления. Развивается заболевание постепенно, в 2/3 случаев в возрасте до 15 лет. В детском возрасте первыми симптомами болезни могут быть нарушение походки, спастическая кривошея; у взрослых чаще встречаются первично-генерализованные формы. В результате нарушения соотношения функции мышц-синергистов и антагонистов возникают насильственные длительные тонические сокращения мышц туловища, головы, тазового пояса, конечностей, обычно ротаторного характера, сочетающиеся с атетоидными движениями в пальцах. Создается впечатление, что мышцы постоянно сокращаются для преодоления действия антагонистов. Возникающие позы, даже самые неудобные, сохраняются в течение длительного времени. Гиперкинезы усиливаются при волнении, активных движениях, во сне исчезают. Постепенно, по мере прогрессирования заболевания, поза пациента становится постоянно дистонической, с усиленным поясничным лордозом, флексией бедер, медиальной ротацией рук и ног. В зависимости от распространенности дистонических явлений выделяют локальную и генерализованную формы заболевания. При локальных дистонических симптомах возникает тоническое сокращение отдельных мышечных групп, нарушаются произвольные движения и возникает аномальная поза. К таким симптомам относятся спастическая кривошея, писчий спазм, оромандибулярная дистония (открывание и закрывание рта и непроизвольные движения языка), блефароспазм, щечно-лицевая, щечно-язычная дистония, хореоатетоз.
Течение и прогноз. Заболевание в большинстве случаев неуклонно прогрессирует. Иногда отмечаются различной длительности ремиссии. Быстро происходит глубокая инвалидизация больных и наступает летальный исход, особенно при генерализованной форме.
Лечение. Длительное, симптоматическое. Применяют комбинации холинолитиков и седативных препаратов, в некоторых случаях эффективно использование леводопы. Назначается также галоперидол или резерпин. Очень редко прибегают к стереотаксическим операциям на подкорковых ядрах.
Семейная атаксия Фридрейха – наследственное дегенеративное заболевание нервной системы, характеризующееся синдромом поражения задних и боковых канатиков спинного мозга. Тип наследования аутосомно-рецессивный, с неполной пенетрантностью патологического гена. Мужчины и женщины болеют одинаково часто.
Патоморфология. Обнаруживаются дегенеративные изменения в проводящих путях задних и боковых канатиков спинного мозга, преимущественно пучков Голля, в меньшей степени – Бурдаха, Флексига, Говерса, волокнах пирамидного пути, задних корешках, а также в клетках коры мозжечка, подкорковых ганглиев, коры большого мозга.
Клинические проявления. Начало заболевания относится к 6—15-летнему возрасту. Первым симптомом болезни является неустойчивая походка, которая была охарактеризована Шарко как табетически-мозжечковая. В ранних стадиях атаксия выражена преимущественно в ногах. По мере прогрессирования заболевания нарушения координации распространяются на верхние конечности и лицо. При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно-суставного чувства и вибрационной чувствительности. Меняется почерк. Ранним симптомом является снижение, а затем угасание сухожильных и периостальных рефлексов. Мышечный тонус понижен. В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей, нередки патологические пирамидные рефлексы, дистальные мышечные атрофии. Интеллект снижен.
Заболевание медленно прогрессирует. Средняя продолжительность жизни 10—15 лет с момента его развития.
Диагностика и дифференциальный диагноз. Заболевание распознается на основании характерных симптомов – деформаций стоп по типу стопы Фридрейха (высокий свод, экстензия основных фаланг пальцев стопы и флексия концевых фаланг), поражения миокарда, эндокринных расстройств.
Дифференцировать заболевание следует от церебрального сифилиса, рассеянного склероза, фуникулярного миелоза и других форм мозжечковых дегенерации.
Лечение. Применяются симптоматические средства: общеукрепляющие препараты, лечебная физкультура, массаж. В некоторых случаях производится хирургическая коррекция деформации стоп.
Мозжечковая атаксия Пьера Мари – наследственное дегенеративное заболевание с преимущественным поражением мозжечка и его проводящих путей. Тип наследования аутосомно-доминантный. Возникает заболевание в возрасте 20 лет и старше.
Патоморфология. Выявляется дегенеративное поражение клеток коры и ядер мозжечка, спиноцеребеллярных путей в боковых канатиках спинного мозга, в ядрах моста и продолговатого мозга.
Клинические проявления. Заболевание проявляется нарушениями функций мозжечка и его связей. Наблюдаются атаксия при выполнении координаторных проб, нарушение походки, скандированная речь, интенционное дрожание, нистагм. Мозжечковые симптомы сочетаются с умеренными или выраженными признаками пирамидной недостаточности (повышение сухожильных и периостальных рефлексов, клонусы стоп), а иногда с глазодвигательными нарушениями (косоглазие, птоз, недостаточность конвергенции). Характерным признаком является в различной степени выраженное снижение интеллекта.
Диагностика и дифференциальный диагноз. Наибольшие трудности возникают при дифференциации наследственной мозжечковой атаксии Пьера Мари и атаксии Фридрейха. Нужно учитывать тип наследования заболевания, возраст, в котором развиваются первые симптомы, характер изменения сухожильных рефлексов (при атаксии Фридрейха они снижены), наличие зрительных и глазодвигательных расстройств при атаксии Пьера Мари, деформации стоп и скелета. Рассеянный склероз в отличие от семейной атаксии Пьера Мари характеризуется ремитирующим течением, большей выраженностью нижнего спастического парапареза, расстройством функций тазовых органов.
Лечение. Симптоматическое.
Группа наследственных заболеваний нервной системы, характеризующихся дегенеративными изменениями нейронов мозжечка, ядер нижних олив и моста мозга, в ряде случаев – ядер черепных нервов каудальной группы, в меньшей степени – поражением проводящих путей и клеток передних рогов спинного мозга, базальных ганглиев. Заболевания отличаются типом наследования и различным сочетанием клинических симптомов. По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенерации.
Тип I – оливопонтоцеребеллярная дегенерация Менделя.Наследуется по аутосомно-доминантному типу. Течение медленно прогрессирующее. Проявляться может в возрасте от 11 до 60 лет. Клиническая картина складывается из симптомов поражения мозжечка (атаксия, мышечная гипотония, скандированная речь с элементами дизартрии, интенционное дрожание), ядер каудальных черепных нервов (дизартрия, дисфагия), подкорковых ганглиев (гиперкинезы); реже выявляются пирамидные и глазодвигательные симптомы.
Тип II – оливопонтоцеребеллярная дегенерация Фиклера—Винклера.Наследуется по аутосомно-рецессивному типу. Проявляется в возрасте от 20 до 80 лет симптомами поражения мозжечка, преимущественно атаксией в конечностях. Чувствительность и сухожильные рефлексы не изменены. Парезов не наблюдается.
Тип III – оливопонтоцеребеллярная дегенерация с ретинальной дегенерацией.Наследуется по аутосомно-доминантному типу. Возникает в молодом возрасте. Наряду с мозжечковыми и экстрапирамидными симптомами определяется прогрессирующее снижение остроты зрения вследствие пигментной дегенерации ганглиозных клеток сетчатки.
Тип IV – оливопонтоцеребеллярная дегенерация Шута—Хайкмана.Наследуется по аутосомно-доминантному типу. Проявляется в детском и молодом возрасте. Кроме мозжечковых симптомов, выявляется поражение ядер VII, IX, Х и XII пар черепных нервов (паралич лицевого нерва, бульбарные симптомы) и задних канатиков спинного мозга (расстройства мышечно-суставного чувства и вибрационной чувствительности).
Тип V – оливопонтоцеребеллярная дегенерация с деменцией, офтальмоплегией и экстрапирамидными нарушениями. Тип наследования аутосомно-доминантный. Развивается в среднем возрасте. Характеризуется деменцией, прогрессирующей офтальмоплегией, экстрапирамидными и мозжечковыми симптомами.
Дифференцировать оливопонтоцеребеллярные дегенерации следует от наследственной атаксии Фридрейха и Пьера Мари, прогрессирующих форм рассеянного склероза, опухолей мозжечка, ювенильных форм паркинсонизма.
Лечение. Симптоматическое. Проводят курсы неспецифического общеукрепляющего лечения, массаж, лечебную физкультуру.
Наследственные нервно-мышечные заболевания(ННМЗ)
Классификация ННМЗ
1. Первичные прогрессирующие мышечные дистрофии (первичные ПМД, миопатии)
2. Вторичные ПМД, или нейрогенные(вторичные) амиотрофии, т.е. спинальные и невральные амиотрофии, обусловленные поражением на различных уровнях (тела или аксона) периферических двигательных нейронов
3. Врожденные непрогрессирующие миопатии
5. Пароксизмальные параличи
Общие признаки ННМЗ:
1.мышечная слабость проявляется симметрично и прогрессирует постепенно
2.мышечная слабость не сопровождается перманентной болью, хотя и возможны болезненные мышечные спазмы – крампи
3.при большинстве форм ПМД слабость раньше проявляется и преобладает в мышцах тазового или плечевого пояса и проксимальных отделах конечностей
4.сухожильные рефлексы снижаются пропорционально выраженности мышечной слабости
5. парестезии, расстройства поверхностной и глубокой чувствительности встречаются нечасто, обычно при невральных амиотрофиях
6. заболевание, как правило, не влияет на функции тазовых органов
ПЕРВИЧНЫЕ ПРОГРЕССИРУЮШИЕ МЫШЕЧНЫЕ ДИСТРОФИИ (МИОПАТИИ)
Относят наследственные болезни, при которых расстройства метаболизма ведут к первичной дистрофии мышц (миопатии)
ХАРАКТЕРНО:
Нарастающая мышечная слабость
Гипотония мышц
Гипотрофия мышц
Сухожильная и периостальная гипорефлексия до арефлексии
Ограничение объема активных движений
Иногда псевдогипертрофия мышц
Отсутствуют фибриллярные и фасцикулярные подергивания
В дебюте чаще страдают мышцы тазового пояса, реже плечевого
Выраженные изменения креатин-креатининового обмена
Снижение механической возбудимости мышц
Миодистрофия псевдогипертрофическая ДЮШЕННА (Х-сцепл. рецессивн. тип)
утрата мышечного белка дистрофина
наиболее злокачественная форма первичных миодистрофий
дебют в раннем детском возрасте
с 2-5 лет уже развивается слабость мышц ТАЗОВОГО пояса, бедер, утиная походка
распространение процесса восходящее (до плечевого пояса)
особенно типична псевдогипертрофия икроножных мышц
со временем возникает слабость мышц лица, языка, глотки, гортани, дыхательных мышц
возможны сухожильные ретракции (чаще пяточного)
развивается кардиопатия
возможен адипозо-генитальный синдром, гипоплазия надпочечников, остеопороз
у 30% отставание в интеллектуальном развитии
высокая степень гиперферментемии (КФК)
Поздняя псевдогипертрофическая Миодистрофия БЕККЕРА-КИНЕРА (Х-сцепл. рецессивн. тип)
дебют от 5 до 20 лет чаще 10-15 лет
течение медленно прогрессирующее
распространение мышечных дистрофий как при миодистрофии Дюшена
поражение сердца выражены меньше
доживают до 30-60 лет, могут иметь детей, интеллект сохранен
повышение активности КФК умеренное
называют мягкой формой миодистрофии Дюшена
качественное изменение белка дистрофина
Миодистрофия ЭМЕРИ-ДРЕЙФУСА-ХОГАНА (Х-сцепл. рецессивн. тип)
дебют с 4-5 летнего возраста
поражение мышц тазового пояса при интактности дистальных отделов конечностей
рано ретракция пяточных сухожилий
псевдогипертрофий нет
позже распространение на плечевой пояс, контрактуры локтевых суставов, др крупн суставов
в поздних стадиях ригидность позвоночника, может быть слабость мышц лица
характерно развитие миокардиодистрофии
интеллект обычно сохранен
иногда доживают до 60 лет
повышение КФК умеренное
ЦЕНТРОНУКЛЕАРНАЯ (миотубулярная) миопатия
форма новорожденных или в период с 5 до 30 лет(поздняя форма)
характерна генерализованная мышечная дистония
в случае поздней формы сначала слабость мышц плечевого и тазового пояса, лица, верхних век, мышц обеспечивающих движения глазных яблок
вытянутый лицевой череп, деформация грудной клетки, Х-образная форма ног
миокардиодистрофия
развитие интеллекта обычное
на ЭМГ изменения первично-мышечного характера
Миодистрофия МЭБРИ (Х-сцепл. рецессивн. тип)
проявляется у мальчиков в пубертатном периоде
слабость мышц тазового пояса и бедер
позже выраженные мышечные псевдогипертрофии
нехарактерны сухожильные контрактуры
кардиомиопатия, липоматоз
интеллект не страдает
течение медленно прогрессирующее
Миодистрофия РОТТАУФА-МОРТЬЕ-БЕЙЕРА (фиброзирующая миопатия) (Х-сцепл. рецессивн. тип)
дебют в детском или юношеском возрасте (чаще 5-12 лет)
выраженные сухожильные ретракции и контрактуры (ограничение тыльн разгиб стоп, затем сгибания шеи, разгибания в локтевых суставах
постепенно формируются патологические позы из-за фиброза мышц
далее невозможно сгибать позвоночник
медленно прогрессируют мышечные гипотрофии
слабость мышц обычно умеренная
преобладают парезы и гипотрофии в лопаточно-плечевой обл. и дистальных отделах ног
псевдогипертрофий нет
характерна кардиомиопатия
интеллект чаще сохранен
выраженная гиперферментемия
нередко доживают до 40-50 лет, умирают от сердечной недостаточности
Ювенильная миодистрофия ЭРБА-РОТА (по аутосомно-рецессивному типу)
дебют в детском или юношеском возрасте чаще в 14-16 лет
конечностно-поясная миодистрофия
прежде всего атрофии мышц тазового пояса
ранний признак утиная походка и др миопатические феномены
в дальнейшем атрофии мышц плечевого пояса, рук (форма ЛЕЙДЕНА-МЕБИУСА)
редко дебют со слабости мышц плечевого пояса (форма ЭРБА)
возможны умеренные псевдогипертрофии, формирование контрактур
при поражении межреберных мышц и диафрагмы – дыхательная недостаточность
мышцы лица чаще не страдают
нередко эндокринопатии
течение вариабельное от мягкого до быстро прогрессирующего
умеренная гиперферментемия
инвалидизация через 10-20 лет
возможна и злокачественная (псевдодюшенновская) форма, дебют в 3-5 лет
Плечелопаточно-лицевая миодистрофия ЛАНДУЗИ-ДЕЖЕРИНА (по аутосомно-рецессивному типу)
дебют чаще к 20 годам, иногда несколько позже
слабость и гипотрофия мышц лица, особенно круговых глаз и рта, мышц плечевого пояса
рано губы тапира, лицо сфинкса, улыбка Джоконды, крыловидные лопатки
далее: слабость передней зубчатой, большой грудной, нижних отделов трапецивидных мышц, широчайшей мышцы спины, двуглавой, трехглавой мышц
далее: слабость перонеальных мышц (появляется степаж)
далее: в меньшей степени проксимальные мышцы нижних конечностей
возможна умеренная псевдогипертрофия икроножных и дельтовидных мышц
сухожильные рефлексы постепенно снижаются
интеллект сохранен
течение относительно мягкое
гиперферментемия умеренная
женщины в 3 раз чаще мужчин болеют
Лопаточно-перонеальная миодистрофия ДАВИДЕНКОВА
1. аутосомно-доминантная форма
проявляется чаще в детстве
иногда во 2-3-м десятилетии жизни
слабость и прогрессирующая гипотрофия мышц плечевого пояса и перонеальной группы
мышц с угасанием сухожильных рефлексов начиная с пяточных, степаж
слабость проксимальных отделов рук и плечевого пояса
возможны дистальные парестезии, гипестезии
как правило не страдают мышцы лица
течение медленно прогрессирующее
возможно развитие мышечных контрактур
повышена активность КФК в крови
2. Х-сцепленная рецессивная форма
дебют в первую декаду жизни иногда с мышечных контрактур
сначала слабость в грудных, дельтовидных мышцах, в мышцах проксимальных отделов рук
позже: перонеальные мышцы
характерно значительное повышение КФК
характерна кардиомиопатия (чаще причина смерти)
Поздняя дистальная миопатия ГОВЕРСА-ВЕЛАНДЕР (миодистрофия ВЕЛАНДЕР) (по аутосомно- доминантный тип с неполн пенетрантностью)
дебют обычно после 20 лет чаще 40-60 лет
медленно прогрессирующее течение
начало со слабости и гипотрофии в мышцах стоп и голеней
позже постепенно мышцы кистей и предплечий
снижаются и исчезают сухожильные и периостальные рефлексы
в поздней стадии поражаются проксимальные мышцы конечностей
чувствительность сохранна
всегда интактны мышцы лица
нет псевдогипертрофий
нехарактерны сухожильные ретракции
возможна кардиомиопатия
Окулярная миодистрофия (наружняя хроническая прогрессирующая офтальмоплегия ГРЕФЕ)
клиника проявляется до 30 лет
медленно нарастающее поражение наружных глазных мышц
протекает, как правило без диплопии и приводит к параличу взора
зрачковые реакции сохранены
первой страдает мышца поднимающая верхнее веко
в далеко зашедшей стадии – двусторонний птоз
со временем могут поражаться мимические, бульбарные, затем скелетные мышцы
Наружная хроническая прогрессирующая офтальмоплегия (миопатия КИЛОХА – НЕВИНА) (по аутосомно- доминантный тип с неполн пенетрантностью)
дебют от 8 мес до 80 лет, чаще на 3-ем десятилетии жизни
медленно нарастающий птоз век, слабость наружных мышц глаза, круговых мышц глаза
(может ассиметрично)
постепенно парез взора вверх, затем в стороны, далее наружная офтальмоплегия
может быть, слабость др мышц лица, жевательных, глотки и гортани (БУЛЬБАРНО-
ОФТАЛЬМОПЛЕГИЧЕСКАЯ форма)
чаще у женщин
Окулофарингеальная миодистрофия (аутосомно-доминантный тип)
прогрессирующая наружная офтальмоплегия + дисфагия и дисфония
1 ВАРИАНТ: характерен птоз век и парез мышц глотки
2 ВАРИАНТ: +парез глазодвигательных мышц, мимических и жевательных мышц,
мышц шеи и проксимальных отделов конечностей
3 ВАРИАНТ: + дистальные отделы конечностей
КФК норма или повышение
ЭМГ признаки миодистрофии
Прогрессирующая офтальмоплегическая миопатия (ядерная с пигментным ретинитом, скротальным языком и снижением интеллекта) проявляется наружным офтальмопарезом/плегией + симптомы по определению
скротальный язык = увеличен с глубокими поперечными бороздами
Окулокраниоскелетный миопатический синдром (синдром КЕРНСА – ШАЯ) (аутосомно-доминантный тип)
дебют до 15 лет в виде наружной офтальмоплегии
далее признаки ПМД на лице, с распространением на мышцы шеи, плечевого и тазового
пояса, проксимальные отделы конечностей
может быть снижение слуха, бульбарный синдром
в ЦСЖ белково-клеточная диссоциация
Поздняя прогрессирующая мышечная дистрофия ШНЕЙДЕРМАНА (аутосомно-доминантный тип)
дебют в 30-40 лет
преимущественно страдают проксимальные отделы конечностей и мышцы лица
прогрессирует медленно
на ЭМГ признаки первичного поражения мышц
Болезнь ВЕРХЕРБЕКА
дебютирует мышечными спазмами в ногах
далее снижение силы в ногах и гипотрофия мышц ног
далее распространение вверх
Мышечная дистрофия с контрактурами ДРЕЙФУСА (по рецессивный, сцепленный с полом тип)
дебют в возрасте 4-5 лет
нарастающая мышечная слабость, преимущественно в мышцах тазового пояса и ног
при ходьбе опирается на большие пальцы стоп
далее формируется поясничный гиперлордоз
особенность: формирование контрактур локтевых и др суставов
псевдогипертрофии нет
нередко поражается миокард, отставание в психическом развитии
Синдром ЛУНДБОРГА (аутосомно-рецессивный тип)
в первые годы жизни олигофрения, катаракта
с пубертатного периода прогрессирующая мышечная слабость преимущественно в прокси-
мальных отделах конечностей
гиперлордоз, утиная походка
половой инфантилизм
со временем слабость мышц шеи, лица, наружных мышц глаза
в поздних стадиях полная обездвиженность
Миопатии митохондриальные
проявляется чаще на 2-м десятилетии жизни
в дебюте: птоз верхних век, наружный офтальмопарез без диплопии (поражение симметр-но)
слабость и похудание сначала в проксимальных мышцах
сухожильная гипорефлексия
длительность прогрессирования процесса вариабельна (мес-десятилетия)
миопатические, невропатические, нервно-мышечные нарушения, вегетативные, обменные и эндокринные расстройства
Читайте также:
