
Генетические заболевания нижних конечностей
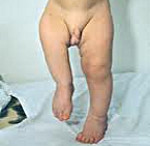
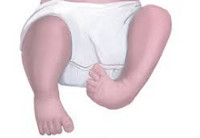
Врожденные аномалии нижних конечностей – обруппа пороков развития, включающая в себя патологию области тазобедренного сустава, бедра, коленного сустава, голени, голеностопного сустава и стопы. Может наблюдаться полное отсутствие конечности или какого-то из ее отделов, недоразвитие целого сегмента или одной из костей, входящих в этот сегмент, недоразвитие мышц, сосудов и нервов, соединительнотканные перетяжки и т. д. Возможно сочетание нескольких врожденных аномалий. Диагноз выставляется на основании данных осмотра, результатов рентгенографии, КТ, МРТ и других исследований. Лечение обычно хирургическое. Прогноз зависит от тяжести патологии.
МКБ-10
- Причины
- Классификация
- Виды аномалий
- Недоразвитие бедренной кости
- Врожденный вывих бедра и дисплазия тазобедренного сустава
- Вальгусная и варусная деформация бедра
- Врожденный вывих надколенника
- Отсутствие надколенника
- Врожденный вывих голени
- Вальгусные и варусные деформации коленного сустава
- Аплазия или недоразвитие большеберцовой кости
- Ложный сустав большеберцовой кости
- Цены на лечение
Общие сведения
Пороки развития нижних конечностей представляют собой обширную группу аномалий, возникших во внутриутробном периоде и значительно различающихся по тяжести. В практике ортопедии и травматологии встречаются относительно часто и составляют 55% от общего количества врожденных аномалий опорно-двигательного аппарата. Грубые пороки диагностируются сразу после рождения. Мелкие аномалии в ряде случаев могут протекать бессимптомно или почти бессимптомно и становиться случайной находкой при обследовании по поводу других травм и заболеваний. Лечением врожденных аномалий нижних конечностей занимаются детские ортопеды и травматологи.
Причины
Аномалии развития могут возникать в результате мутаций, а также действия внешних и внутренних тератогенных факторов. Наиболее распространенными внешними факторами, оказывающими негативное влияние на формирование конечностей, являются инфекционные болезни, нарушения питания, прием ряда медикаментов и радиационные воздействия. К числу внутренних факторов, которые могут привести к нарушению формирования конечностей, относится пожилой возраст матери, патология матки, тяжелые соматические болезни, некоторые гинекологические заболевания и эндокринные нарушения.
Классификация
Пороки, которые возникают вследствие недостаточности формирования:
- Амелия – конечность полностью отсутствует. Возможен апус (отсутствуют обе нижние конечности) и монопус (отсутствует одна нижняя конечность).
- Фокомелия или тюленеобразная конечность. Отсутствуют средние и/или проксимальные части конечности вместе с соответствующими суставами. Может быть двухсторонней или односторонней. Иногда в процесс вовлекаются все конечности, как нижние, так и верхние. Полная фокомелия – голень и бедро отсутствуют, сформированная стопа прикрепляется к туловищу. Дистальная фокомелия – голень отсутствует, стопа прикрепляется к бедру. Проксимальная фокомелия – бедро отсутствует, голень со стопой прикрепляются к туловищу.
- Перомелия – разновидность фокомелии, при которой наблюдается отсутствие части конечности в сочетании с недоразвитием ее дистального отдела (стопы). Полная перомелия – нога отсутствует, в месте ее прикрепления располагается кожный выступ или рудиментарный палец. Неполная – бедро отсутствует или недоразвито, конечность также заканчивается кожным выступом или рудиментарным пальцем.
Кроме того, различают отсутствие (аплазию) малоберцовой или большеберцовой кости, отсутствие фаланг (афалангию), отсутствие пальцев (адактилию), наличие одного пальца на стопе (монодактилию), а также типичную и атипичную формы расщепления стопы из-за отсутствия или недоразвития ее средних отделов.
Пороки, которые возникают вследствие недостаточной дифференцировки:
Сиреномелия – слияние нижних конечностей. Может наблюдаться слияние только мягких тканей либо слияние как мягких тканей, так и некоторых трубчатых костей. Нередко сочетается с отсутствием или недоразвитием костей таза и конечностей. При сиреномелии возможно как отсутствие стоп, так и наличие одной или двух стоп (чаще рудиментарных). Обычно наблюдается одновременное недоразвитие прямой кишки, заднего прохода, мочевой системы, внутренних и наружных половых органов.
Кроме того, в группу пороков, обусловленных недостаточной дифференцировкой, включают синостозы (сращения костей), врожденный вывих бедра, врожденную косолапость, артрогрипоз и некоторые другие аномалии.
Пороки вследствие увеличения количества: увеличение количества нижних конечностей – полимелия, удвоение стопы – диплоподия.
Пороки вследствие недостаточного роста – гипоплазии различных костей нижних конечностей.
Пороки вследствие избыточного роста – гигантизм, возникающий при одностороннем увеличении развитой конечности.
Врожденные перетяжки – тканевые тяжи, возникающие в различных местах конечности и зачастую нарушающие ее функцию.
Виды аномалий
Составляет 1,2% от общего количества врожденных деформаций скелета. Часто сочетается с другими аномалиями, в том числе аплазией малоберцовой кости и отсутствием надколенника. Проявляются хромотой. Степень нарушения функции конечности зависит от величины укорочения и тяжести порока развития. При поражении диафиза смежные суставы, как правило, не изменены, их функция сохранена в полном объеме. При поражении дистальных отделов бедра обычно возникают контрактуры. Конечность ротирована, укорочена. Таз перекошен и опущен в сторону дефекта. Ягодичная складка сглажена или отсутствует. Мышцы ягодицы и бедра атрофированы, стопа в положении эквинуса. Рентгенография бедренной кости свидетельствует об укорочении и недоразвитии сегмента.
Лечение хирургическое, направленное на восстановление длины конечности. В раннем возрасте выполняют операции для стимуляции ростковых зон. Начиная с 4-5 лет, осуществляют остеотомию в сочетании с наложением дистракционного аппарата. Если укорочение настолько велико, что восстановление длины конечности не представляется возможным, необходима ампутация стопы, иногда – в сочетании с артродезом коленного сустава (для создания длинной функциональной культи). При незначительном укорочении возможно использование специальной обуви и различных ортопедических аппаратов.
Врожденный вывих бедра наблюдается относительно редко, обычно обнаруживаются различные степени дисплазии. Патология, как правило, односторонняя. Девочки страдают в 7 раз чаще мальчиков. В 5% случаев выявляется прямая передача порока по наследству. Проявляется хромотой, ротацией и укорочением конечности. При двухсторонней аномалии возникает утиная походка. Рентгенография тазобедренного сустава свидетельствует об уменьшении и уплощении головки и ее стоянии выше вертлужной впадины. Лечение в раннем возрасте консервативное с использованием различных аппаратов, специальных трусиков и подушечек. При неустранимых вывихах по достижении 2-3 лет проводится операция.
Развивается при нарушении оссификации шейки или внутриутробном повреждении хряща, одинаково часто наблюдается у девочек и мальчиков, в 30% выявляется с двух сторон. Вальгусная деформация, как правило, протекает бессимптомно. Варусное искривление сопровождается хромотой, ограничением движений и быстрой утомляемостью конечности. Клинические проявления напоминают врожденный вывих бедра. При рентгенографии определяется задержка окостенения головки, укорочение и истончение бедренной кости. Шеечно-диафизарный угол уменьшен. Лечение хирургическое, выполняется корригирующая остеотомия для увеличения шеечно-диафизарного угла.
Встречается достаточно редко. Имеется наследственная предрасположенность. Может сочетаться с другими пороками, мальчики страдают вдвое чаще девочек. Врожденный вывих надколенника проявляется быстрой утомляемостью конечности, неустойчивой походкой и частыми падениями. Возможна контрактура. Без лечения проблема с возрастом усугубляется, возникает деформирующий артроз, развивается вальгусное искривление конечности. Рентгенография коленного сустава свидетельствует о недоразвитии и смещении надколенника (чаще кнаружи) и недоразвитии наружного мыщелка. Лечение хирургическое – собственную связку надколенника перемещают и фиксируют в срединном положении.
Часто сочетается с другими аномалиями развития коленного сустава (недоразвитием суставных концов большеберцовой и бедренной кости), с вывихом бедра и голени, косолапостью и прочими пороками. Течение изолированной патологии обычно бессимптомное, при повышенных нагрузках возможна слабость и утомляемость конечности. При изолированной аномалии лечение не требуется.
Выявляется редко, обычно носит двухсторонний характер. Сопровождается контрактурой и деформацией колена. Тип деформации зависит от вида смещения костей голени. Мышцы бедра и голени недоразвиты, часто имеют аномальные точки прикрепления. Патология нередко сочетается с аномалиями развития голеностопного сустава, отсутствием или недоразвитием большеберцовой кости. Лечение в раннем возрасте консервативное (вытяжение с последующим вправлением). В возрасте 2 года и старше проводятся операции – открытое вправление вывиха, при необходимости в сочетании с коррекцией сопутствующей скелетной патологии.
Наблюдаются редко, могут передаваться по наследству. Обычно сочетаются с деформацией шейки бедра и плоскостопием. Становятся причиной раннего тяжелого гонартроза. В возрасте до 5-6 лет проводится коррекция с использованием консервативных методов, в последующем осуществляется оперативное вмешательство. В зависимости от тяжести патологии проводят изолированную остеотомию в области надмыщелков бедренной кости либо сочетают остеотомию бедра с желобковой, клиновидной или поперечной остеотомией большеберцовой кости.
Сопровождается укорочением и искривлением конечности. Стопа супинирована, находится в положении эквинуса или подвывиха. Опора нарушена. Возможно сочетание с недоразвитием или отсутствием костей стопы, недоразвитием или вывихом надколенника, атрофией и нарушением развития мышц голени и бедра. Детям до 3 лет проводится консервативная терапия для восстановления нормального положения стопы. В последующем выполняется удлинение голени с использованием дистракционных аппаратов.
Может быть истинным или возникшим в месте расположения врожденной кисты. Выявляется патологическая подвижность, углообразное или дугообразное искривление в области ложного сустава, атрофия мышц, уплотнение и рубцовые изменения кожи, укорочение и утончение конечности. Рентгенография костей голени свидетельствует об остеопорозе. Лечение хирургическое с использованием костных трансплантатов или аппарата Илизарова.
24.1. Нервно-мышечные заболевания
Наследственные нервно-мышечные заболевания – большая гетерогенная группа болезней, в основе которых лежит генетически детерминированное поражение нервно-мышечного аппарата. Заболевания характеризуются мышечной слабостью, мышечными атрофиями, нарушениями статических и локомоторных функций.
При постановке диагноза учитываются возраст проявления первых клинических симптомов заболевания, локализация атрофии и характер распространения миодистрофического процесса (восходящий, нисходящий, наличие или отсутствие псевдогипертрофий, фасцикуляций, расстройств чувствительности, пароксизмов мышечной слабости), а также темп течения.
Прогрессирующие мышечные дистрофии – наиболее обширная группа. В зависимости от характера первичных изменений условно различают первичные (миопатии) и вторичные формы прогрессирующих мышечных дистрофий (денервационные амиотрофии – спинальные и невральные).
Наследственные пароксизмальные миоплегии – группа нервно-мышечных заболеваний, характеризующихся внезапными приступами мышечной слабости и плегиями. Наиболее распространенными из наследственных пароксизмальных миоплегии являются гипо-, гипер– и нормокалиемическая формы. Патогенез неясен. Предполагается генетически детерминированный дефект мембраны сарколеммы, нарушающий проницаемость для ионов натрия и калия,
Гипокалиемическая форма пароксизмальной миоплегии (болезнь Вестфаля). Заболевание описано Вестфалем в 1895 г. Наследуется по аутосомно-доминантному типу.
Клинические проявления. Болезнь проявляется в возрасте 6—15 лет. Пароксизмы характеризуются внезапным в ночные или утренние часы развитием мышечной слабости, обездвиженности, снижением мышечного тонуса, сухожильных рефлексов, вегетативными расстройствами – лабильностью пульса, артериального давления, гипергидрозом. Приступы бывают парциальными, охватывающими небольшую группу мышц, и генерализованными. Во время приступа возникают нарушения сердечно-сосудистой деятельности: систолический шум, изменения ЭКГ. Сознание всегда сохранено. Средняя продолжительность приступа – несколько часов, крайне редко пароксизмы держатся несколько суток. Содержание калия в крови во время приступа менее 2 ммоль/л и ниже. Частота приступов вариабельна. Они провоцируются перееданием пищи, богатой углеводами, охлаждением, физическими нагрузками.
Лечение. Диета, богатая калием (чернослив, курага, картофель, изюм). Для купирования приступа назначают 10% раствор хлорида калия внутрь (по 1 столовой ложке каждый час) или 0,5% раствор в изотоническом растворе хлорида натрия внутривенно (2—2,5 г на 500 мл раствора в течение часа). Целесообразно применять также панангин внутривенно капельно.
Гиперкалиемическая форма пароксизмальной миоплегин (болезнь Гамсторп). Заболевание описано И.Гамсторп в 1956 г. Наследуется по аутссомно-доминантному типу.
Клинические проявления. Болезнь проявляется в возрасте 1—5 лет. Симптоматика сходна с пароксизмами при гипокалиемической форме и характеризуется внезапным развитием мышечной слабости, плегиями, снижением мышечного тонуса, сухожильных рефлексов, вегетативными расстройствами. В отличие от гипокалиемического гиперкалиемический паралич развивается обычно днем, сопровождается выраженными парестезиями, сочетается со слабостью мышц лица, артикуляционного аппарата, имеет меньшую продолжительность (30—40 мин). Во время приступа содержание калия в крови повышается до 6—7 ммоль/л. Частота приступов вариабельна: от ежедневных до нескольких раз в месяц. В межприступные периоды неврологическая симптоматика отсутствует. Провоцирующими факторами являются голодание, физические нагрузки, вызывающие утомление.
Лечение. Диета с повышенным содержанием углеводов, поваренной соли, ограниченным количеством калия. Вводят 40 мл 40% раствора глюкозы внутривенно вместе с инсулином подкожно; 20 мл 10% раствора хлорида кальция внутривенно.
Нормокалиемический (периодический) паралич. Наследуется по аутосомно-доминантному типу.
Клинические проявления. Болезнь проявляется до 10-летнего возраста. Особенностью ее является сравнительно медленно (в течение нескольких суток) пароксизмально нарастающая умеренная слабость в мышцах туловища, конечностей и в жевательной мускулатуре, а также медленный (1—2 нед) регресс симптоматики. Провоцирующими факторами являются продолжительный сон, длительное пребывание в одной позе, переохлаждение.
Лечение. Диета, богатая поваренной солью. Назначают ацетазоламид (диакарб).
Течение. Все формы пароксизмальных миоплегий медленно прогрессируют. Прогноз при своевременно поставленном диагнозе, проведении экстренных мероприятий и дифференцированной медикаментозной терапии благоприятный.
Диагностика и дифференциальный диагноз. Диагноз строится на основании генеалогического анализа, особенностей клинической картины, с учетом возраста, в котором начинается заболевание, времени возникновения пароксизма (ночью, утром, днем, в неопределенное время), степени выраженности мышечной слабости, частоты и длительности приступа, провоцирующих факторов, данных лабораторного биохимического исследования (содержание биоэлектрической активности мышц).
Дифференцировать заболевание следует от миоплегий, развивающихся в результате первичных эндокринных заболеваний, – тиреотоксикоза, болезни Конна (первичный гиперальдостеронизм), болезни Аддисона и др.
Лечение. Показана диета, богатая поваренной солью. Назначают диакарб.
24.2. Пирамидные и экстрапирамидные дегенерации
Хроническое прогрессирующее заболевание нервной системы, клинически проявляющееся изменениями мышечного тонуса и непроизвольными тоническими сокращениями мышц туловища и конечностей.
Этиология и патогенез. Различают идиопатическую (семейную) торсионную и симптоматическую дистонию. Тип наследования при идиопатической торсионной дистонии как аутосомно-доминантный, так и аутосомно-рецессивный. Симптоматическая торсионная дистония встречается при гепатоцеребральной дистрофии, хорее Гентингтона, опухолях мозга, эпидемическом энцефалите, детском церебральном параличе. Имеются указания, что в патогенезе наследственной торсионной дистонии имеет значение нарушение допаминового обмена. При обследовании у этих больных обнаруживается повышение содержания допамин-?-гидроксилазы в сыворотке крови.
Патоморфология. Дистрофические изменения обнаруживаются преимущественно в мелких нейронах в области скорлупы чечевицеобразного ядра, реже – в других базальных ганглиях.
Клинические проявления. Развивается заболевание постепенно, в 2/3 случаев в возрасте до 15 лет. В детском возрасте первыми симптомами болезни могут быть нарушение походки, спастическая кривошея; у взрослых чаще встречаются первично-генерализованные формы. В результате нарушения соотношения функции мышц-синергистов и антагонистов возникают насильственные длительные тонические сокращения мышц туловища, головы, тазового пояса, конечностей, обычно ротаторного характера, сочетающиеся с атетоидными движениями в пальцах. Создается впечатление, что мышцы постоянно сокращаются для преодоления действия антагонистов. Возникающие позы, даже самые неудобные, сохраняются в течение длительного времени. Гиперкинезы усиливаются при волнении, активных движениях, во сне исчезают. Постепенно, по мере прогрессирования заболевания, поза пациента становится постоянно дистонической, с усиленным поясничным лордозом, флексией бедер, медиальной ротацией рук и ног. В зависимости от распространенности дистонических явлений выделяют локальную и генерализованную формы заболевания. При локальных дистонических симптомах возникает тоническое сокращение отдельных мышечных групп, нарушаются произвольные движения и возникает аномальная поза. К таким симптомам относятся спастическая кривошея, писчий спазм, оромандибулярная дистония (открывание и закрывание рта и непроизвольные движения языка), блефароспазм, щечно-лицевая, щечно-язычная дистония, хореоатетоз.
Течение и прогноз. Заболевание в большинстве случаев неуклонно прогрессирует. Иногда отмечаются различной длительности ремиссии. Быстро происходит глубокая инвалидизация больных и наступает летальный исход, особенно при генерализованной форме.
Лечение. Длительное, симптоматическое. Применяют комбинации холинолитиков и седативных препаратов, в некоторых случаях эффективно использование леводопы. Назначается также галоперидол или резерпин. Очень редко прибегают к стереотаксическим операциям на подкорковых ядрах.
Семейная атаксия Фридрейха – наследственное дегенеративное заболевание нервной системы, характеризующееся синдромом поражения задних и боковых канатиков спинного мозга. Тип наследования аутосомно-рецессивный, с неполной пенетрантностью патологического гена. Мужчины и женщины болеют одинаково часто.
Патоморфология. Обнаруживаются дегенеративные изменения в проводящих путях задних и боковых канатиков спинного мозга, преимущественно пучков Голля, в меньшей степени – Бурдаха, Флексига, Говерса, волокнах пирамидного пути, задних корешках, а также в клетках коры мозжечка, подкорковых ганглиев, коры большого мозга.
Клинические проявления. Начало заболевания относится к 6—15-летнему возрасту. Первым симптомом болезни является неустойчивая походка, которая была охарактеризована Шарко как табетически-мозжечковая. В ранних стадиях атаксия выражена преимущественно в ногах. По мере прогрессирования заболевания нарушения координации распространяются на верхние конечности и лицо. При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно-суставного чувства и вибрационной чувствительности. Меняется почерк. Ранним симптомом является снижение, а затем угасание сухожильных и периостальных рефлексов. Мышечный тонус понижен. В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей, нередки патологические пирамидные рефлексы, дистальные мышечные атрофии. Интеллект снижен.
Заболевание медленно прогрессирует. Средняя продолжительность жизни 10—15 лет с момента его развития.
Диагностика и дифференциальный диагноз. Заболевание распознается на основании характерных симптомов – деформаций стоп по типу стопы Фридрейха (высокий свод, экстензия основных фаланг пальцев стопы и флексия концевых фаланг), поражения миокарда, эндокринных расстройств.
Дифференцировать заболевание следует от церебрального сифилиса, рассеянного склероза, фуникулярного миелоза и других форм мозжечковых дегенерации.
Лечение. Применяются симптоматические средства: общеукрепляющие препараты, лечебная физкультура, массаж. В некоторых случаях производится хирургическая коррекция деформации стоп.
Мозжечковая атаксия Пьера Мари – наследственное дегенеративное заболевание с преимущественным поражением мозжечка и его проводящих путей. Тип наследования аутосомно-доминантный. Возникает заболевание в возрасте 20 лет и старше.
Патоморфология. Выявляется дегенеративное поражение клеток коры и ядер мозжечка, спиноцеребеллярных путей в боковых канатиках спинного мозга, в ядрах моста и продолговатого мозга.
Клинические проявления. Заболевание проявляется нарушениями функций мозжечка и его связей. Наблюдаются атаксия при выполнении координаторных проб, нарушение походки, скандированная речь, интенционное дрожание, нистагм. Мозжечковые симптомы сочетаются с умеренными или выраженными признаками пирамидной недостаточности (повышение сухожильных и периостальных рефлексов, клонусы стоп), а иногда с глазодвигательными нарушениями (косоглазие, птоз, недостаточность конвергенции). Характерным признаком является в различной степени выраженное снижение интеллекта.
Диагностика и дифференциальный диагноз. Наибольшие трудности возникают при дифференциации наследственной мозжечковой атаксии Пьера Мари и атаксии Фридрейха. Нужно учитывать тип наследования заболевания, возраст, в котором развиваются первые симптомы, характер изменения сухожильных рефлексов (при атаксии Фридрейха они снижены), наличие зрительных и глазодвигательных расстройств при атаксии Пьера Мари, деформации стоп и скелета. Рассеянный склероз в отличие от семейной атаксии Пьера Мари характеризуется ремитирующим течением, большей выраженностью нижнего спастического парапареза, расстройством функций тазовых органов.
Лечение. Симптоматическое.
Группа наследственных заболеваний нервной системы, характеризующихся дегенеративными изменениями нейронов мозжечка, ядер нижних олив и моста мозга, в ряде случаев – ядер черепных нервов каудальной группы, в меньшей степени – поражением проводящих путей и клеток передних рогов спинного мозга, базальных ганглиев. Заболевания отличаются типом наследования и различным сочетанием клинических симптомов. По классификации Кенигсмарка и Вайнера различают 5 типов оливопонтоцеребеллярных дегенерации.
Тип I – оливопонтоцеребеллярная дегенерация Менделя.Наследуется по аутосомно-доминантному типу. Течение медленно прогрессирующее. Проявляться может в возрасте от 11 до 60 лет. Клиническая картина складывается из симптомов поражения мозжечка (атаксия, мышечная гипотония, скандированная речь с элементами дизартрии, интенционное дрожание), ядер каудальных черепных нервов (дизартрия, дисфагия), подкорковых ганглиев (гиперкинезы); реже выявляются пирамидные и глазодвигательные симптомы.
Тип II – оливопонтоцеребеллярная дегенерация Фиклера—Винклера.Наследуется по аутосомно-рецессивному типу. Проявляется в возрасте от 20 до 80 лет симптомами поражения мозжечка, преимущественно атаксией в конечностях. Чувствительность и сухожильные рефлексы не изменены. Парезов не наблюдается.
Тип III – оливопонтоцеребеллярная дегенерация с ретинальной дегенерацией.Наследуется по аутосомно-доминантному типу. Возникает в молодом возрасте. Наряду с мозжечковыми и экстрапирамидными симптомами определяется прогрессирующее снижение остроты зрения вследствие пигментной дегенерации ганглиозных клеток сетчатки.
Тип IV – оливопонтоцеребеллярная дегенерация Шута—Хайкмана.Наследуется по аутосомно-доминантному типу. Проявляется в детском и молодом возрасте. Кроме мозжечковых симптомов, выявляется поражение ядер VII, IX, Х и XII пар черепных нервов (паралич лицевого нерва, бульбарные симптомы) и задних канатиков спинного мозга (расстройства мышечно-суставного чувства и вибрационной чувствительности).
Тип V – оливопонтоцеребеллярная дегенерация с деменцией, офтальмоплегией и экстрапирамидными нарушениями. Тип наследования аутосомно-доминантный. Развивается в среднем возрасте. Характеризуется деменцией, прогрессирующей офтальмоплегией, экстрапирамидными и мозжечковыми симптомами.
Дифференцировать оливопонтоцеребеллярные дегенерации следует от наследственной атаксии Фридрейха и Пьера Мари, прогрессирующих форм рассеянного склероза, опухолей мозжечка, ювенильных форм паркинсонизма.
Лечение. Симптоматическое. Проводят курсы неспецифического общеукрепляющего лечения, массаж, лечебную физкультуру.
Болезнь Брутона
Болезнь Брутона (первичная гипо- или агаммаглобулинемия взрослых) — наследственное заболевание семейного характера, сцепленное с Х-хромосомой, однако нередко также рецессивное наследование. Часто уже в раннем детстве отмечается склонность к повторным бактериальным инфекциям различной локализации (легкие, придаточные пазухи носа, кожа). К типичным признакам относятся диарея, а также увеличение периферических лимфатических узлов и селезенки. Возможны системные ревматические проявления по типу диффузных болезней соединительной ткани. Суставной синдром характеризуется эпизодической мигрирующей полиартралгией либо острым, подострым, но чаще хроническим моно- или асимметричным олигоартритом крупных суставов. В синовиальной жидкости признаки воспаления слабо выражены или отсутствуют вовсе. Даже при длительном течении артрит не приводит к рентгенологическим изменениям пораженных суставов. В анализах крови СОЭ в норме, острофазовые показатели не изменены, однако снижены уровень гамма-глобулинов (или они отсутствуют), иммуноглобулинов (тотально или избирательно), отсутствуют изогемагглютинины. В костном мозге выявляется отсутствие или снижение содержания плазмоцитов, а в биоптате лимфатического узла — сужение кортикального слоя, первичные фолликулы в нем редкие и малоразвитые.
Болезнь Волкова
Болезнь Гоше
Болезнь Гоше — относительно редкое заболевание, распространенное преимущественно среди евреев-ашкенази и обусловленное дефицитом лизосомальной бета-глюкоцереброзидазы. В результате происходит массивное накопление глюкоцереброзида в селезенке, печени, легких, костном мозге и других органах и тканях с образованием характерных гигантских клеток, содержащих вакуоли глюкоцереброзида (клетки Гоше). У больных, как правило, увеличены печень и селезенка, а иногда и периферические лимфатические узлы. Кроме того, выявляется очаговая кожная пигментация охряного или коричневого цвета, а у некоторых больных — желтые пятна на склерах. У детей наблюдается поражение нервной системы. В анализах крови отмечаются анемия, лейкопения и тромбоцитопения, которая может приводить к геморрагическому синдрому с носовыми кровотечениями и подкожными кровоизлияниями. В ряде случаев обнаруживается моноклоновая гаммапатия. Нередко развиваются костные повреждения (чаще всего деструкция шейки, головки или верхней трети бедра), приводящие к болям, деформации костей, патологическим переломам. Рентгенологическое исследование обнаруживает увеличение объема кости, чередование в ней участков уплотнения и разрежения либо диффузную декальцификацию костей с явлениями спонгиоза, истончение кортикального слоя, кистозные и остеосклеротические изменения. Могут возникать боли и припухлость крупных суставов, чаще нижних конечностей. Иногда поражается также поясничный отдел позвоночника, что приводит к сплющиванию (компрессии) тел позвонков. Для детской формы болезни Гоше типична быстрая смерть, при хроническом течении — медленное прогрессиро-вание. Диагноз основывается на выявлении в стернальном пунктате гигантских клеток Гоше.
Болезнь Мухи—Хаберманна
Болезнь Мухи—Хаберманна (Mucha—Habermann) — довольно распространенное, но редко распознаваемое кожное заболевание, встречающееся у детей. Характерно появление оспоподобных везикул на коже туловища, бедер, предплечий. Эти элементы могут эволюционировать в геморрагии, некроти-зироваться и разрешаются обычно с образованием корочек. Как правило, кожные поражения протекают изолированно, но иногда сопровождаются субфебрильной лихорадкой, а в ряде случаев — артритом (в том числе по типу ревматоидного артрита), склеродермией, интерстициальным пневмонитом, повышенной чувствительностью к укусу пчел. Предполагают возможную связь заболевания с реактивацией инфекции, вызванной вирусом Эпштейна—Барр.
Болезнь Тиманна
Болезнь Тиманна (Thiemann) — наследственное заболевание (аутосомно-доминантный тип наследования), встречающееся у юношей в начальном периоде полового созревания и проявляющееся множественной остеохондропа-тией (асептическим остеонекрозом) фаланг пальцев кистей, а иногда и стоп. Поражаются эпифизы средних фаланг указательного, среднего и безымянного пальцев (кроме большого пальца), обычно вовлекаются одновременно 2— 3 пальца обеих кистей, но не симметрично. Выявляется веретенообразная припухлость в области проксимальных, а иногда и дистальных межфаланго-вых суставов. Могут поражаться также межфаланговый сустав I пальца стоп и I тарзо-метатарзальный сустав. При рентгенологическом исследовании отмечается уменьшение, а нередко резорбция пораженных эпифизов, соответствующая фаланга укорачивается. Возможно спонтанное выздоровление, однако чаще остаются явления выраженного вторичного остеоартроза.
Болезнь Фабри
Болезнь Фабри (Fabry) — врожденное заболевание, характеризующееся наследственным дефицитом фермента альфа-Г4-галактозидазы, приводящим к накоплению гликолипидов (церамида) в цитоплазме и лизосомах клеток различных органов и тканей. Заболевание начинается с детства. Изменения кожи проявляются ее утолщением и ангиоэктазиями, поражение глаз — помутнением роговицы. При поражении синовиальной оболочки суставов происходит утолщение капсулы суставов и сухожильных влагалищ, отмечаются жгучие боли в пальцах кистей и стоп, голенях, предплечьях при повышении внешней температуры и ослабление болей при охлаждении конечностей. Периодически наблюдается отечность голеностопных и коленных суставов на фоне немотивированного субфебрилитета. Таким образом, клиническая картина часто напоминает воспалительное ревматическое заболевание. Рентгенологическое исследование выявляет в костях кистей множественные энтезопатические кальцификаты, а также мелкие интра- и экстраартикулярные эрозии.
Болезнь Файербанка
Болезнь Файербанка (множественная эпифизарная дисплазия) — наследственное заболевание, характеризующееся нарушением развития эпифизов, главным образом длинных трубчатых костей. Заболевание проявляется в детском и юношеском возрасте. У больных отмечаются низкий рост, нарушение походки, контрактуры и деформации преимущественно суставов нижних конечностей, широкие кисти с утолщенными и укороченными пальцами, вывихи и подвывихи в коленном и локтевом суставах, деформация стоп за счет укорочения пальцев. При рентгенологическом исследовании выявляются уменьшение размеров поперечника эпифизов, их деформация, уплощение, нередко — дольчатое строение (фрагментация). Указанные изменения носят симметричный характер и наиболее выражены главным образом в тазобедренных, коленных, плечевых и локтевых суставах.
Мукополисахаридозы
Мукополисахаридозы — группа наследственных заболеваний (аутосомно-рецессивный тип наследования), в основе которых врожденный дефект энзимов, принимающих участие в обмене глюкозаминогликанов — основных компонентов органического матрикса соединительной ткани. Отмечаются аномальная продукция, чрезмерное накопление и выделение одного или нескольких определенных мукополисахаридов, вследствие чего развиваются патологические изменения в хряще (множественный дизостоз), фасциях, периосте, сухожилиях, сердечных клапанах (сморщиваются в результате рубцовых процессов), кровеносных сосудах, черепно-мозговых оболочках, роговице, печени, селезенке (см. синдромы Марото-Лами, Моркио, Пфаундлера-Гурлера, Шийана).
Синдром Джеккейя
Синдром Джеккейя (Giaccai) — редкое семейное заболевание с аутосомно-рецессивным типом наследования, наблюдающееся в районах с высокой частотой близкородственных браков и характеризующееся прогрессирующим симметричным лизисом дистальных частей конечностей (акроостеолизом) с их последующей деформацией. Болезнь начинается в юношеском, реже — в детском возрасте и клинически похожа на остеомиелит и саркому. У больных наблюдается симметричный лизис костей дистальных отделов конечностей и последующая их деформация. На подошвенной поверхности стоп над участком рассасывания костей образуются очаги безболезненной припухлости, чаще без кожной гипертермии. Через некоторое время на этих участках появляются длительно не заживающие кожные дефекты (язвы), через которые происходит спонтанное отторжение мелких костных секвестров. После этого язвенные дефекты заживают, но могут наблюдаться и торпидно текущие.
Течение заболевания рецидивирующее с ремиссиями от нескольких недель до 3—4 лет. В итоге происходит выраженная деформация стоп с ампутацией отдельных ногтевых фаланг, деформацией и укорочением пальцев. Кроме того, отмечаются трофические изменения ногтей, гипотрофия мышц голеней и омозолелость подошвенных поверхностей, гипостезия кистей и стоп с сохранением тактильной чувствительности. Рентгенологически выявляются остеолиз фаланг и дистальных отделов плюсневых костей, очаги их деструкции без четкой периостальной реакции.
Синдром Клиппела—Тренонея
Синдром Клиппела—Тренонея — врожденная сосудистая аномалия в виде обширных варикозных расширений в области конечностей (в том числе кистей и пальцев) и верхнего плечевого пояса. Постепенно развивается гипертрофия мягких тканей конечностей, отмечается тенденция к изъязвлению кожных покровов. Происходит деформация костей (искривление, утолщение) и суставов. Возможно образование анкилозов. В области пораженных суставов могут прощупываться и рентгенологически определяться флеболиты.
Синдром Кускоквим
Синдром Кускоквим (по названию реки в штате Аляска, США, в районе дельты которой впервые были выявлены больные с данным синдромом) — комплекс наследственных аномалий (аутосомно-рецессивный тип наследования), включающий множественные контрактуры крупных суставов (преимущественно коленных и локтевых), косолапость и косорукость.
Синдром Лери
Синдром Лери — наследственное заболевание костей, характеризующееся поражением обычно одной из верхних или нижних конечностей и проявляющееся болями, распространяющимися вниз по конечности, а позже — ограничением подвижности в суставах на пораженной стороне. Часто на соответствующей стороне наблюдаются также склеродермия, атрофия и кальциноз мягких тканей. Рентгенологически обнаруживается эндостальный и частично пе-риостальный остеосклероз, возможно выявление продольных полос кальциноза в мягких тканях пораженной конечности.
Синдром Лутца—Жансельма
Синдром Лутца—Жансельма (Lutz—Jeanselme) — гиперэозинофилия в сочетании с образованием околосуставных узелков. Постепенно появляются различной величины и консистенции безболезненные подкожные узелки, локализующиеся преимущественно над костными выступами на поверхности суставов, чаще на локтях, над мелкими суставами пальцев, над областью тазобедренных суставов. В области мелких суставов кожа над узелками нормального вида, а над крупными суставами — натянутая, синюшная. Характерны артрал-гия, перемежающаяся лихорадка, слабость, кахексия. Возможно развитие неэрозивного полиартрита с вовлечением периартикулярных тканей. В крови и синовиальном выпоте определяется высокая эозинофилия. В анамнезе у больных различные аллергические заболевания. Болезнь тянется годами, обострения чередуются с частыми периодами ремиссии.
Читайте также:
