
Заболевание характеризующиеся повышенной ломкостью костей

ИГА (Итоговая государственная аттестация)
Дисциплина: Гериатрия
№ 1
* 1 -один правильный ответ
Заболевания лиц пожилого и старческого возраста изучает
1) геронтология
2) гериатрия
3) герогигиена
! 2
№ 2
* 1 -один правильный ответ
Наличие двух и более заболеваний у пациента — это
1) полипрагмазия
2) полиморбидность
3) полиэтиологичность
4) атипичность
! 2
№ 3
* 1 -один правильный ответ
Одновременное назначение нескольких лекарственных препаратов — это
1) полипрагмазия
2) полиморбидность
3) полиэтиологичность
4) полиморфность
! 1
№ 4
* 1 -один правильный ответ
При бессоннице лицам пожилого и старческого возраста рекомендуют
1) настой пустырника
2) бромиды
3) барбамил
4) фенобарбитал
! 1
№ 5
* 1 -один правильный ответ
Рентгенографию следует обязательно провести при появлении на фоне хронического бронхита
1) общей слабости
2) недомогания
3) влажного кашля
4) кровохарканья
! 4
№ 6
* 1 -один правильный ответ
Наиболее частое осложнение острого бронхита у лиц пожилого и старческого возраста
1) хронический бронхит
2) очаговая пневмония
3) туберкулез
4) рак легкого
! 2
№ 7
* 1 -один правильный ответ
При возникновении пневмонии у лиц пожилого и старческого возраста редко наблюдается
1) слабость
2) недомогание
3) кашель
4) высокая лихорадка
! 4
№ 8
* 1 -один правильный ответ
К развитию пневмонии у лиц пожилого и старческого возраста предрасполагает
1) переутомление
2) перегревание
3) постельный режим
4) стрессы
! 3
№ 9
* 1 -один правильный ответ
Причина снижения интенсивности боли при инфаркте миокарда у лиц пожилого и старческого возраста
1) усиление воспалительной реакции
2) ослабление воспалительной реакции
3) повышение порога болевой чувствительности
4) снижение порога болевой чувствительности
! 3
№ 10
* 1 -один правильный ответ
Форма инфаркта миокарда, реже встречающаяся в пожилом и старческом возрасте
1) ангинозная
2) астматическая
3) аритмическая
4) безболевая
! 1
№ 11
* 1 -один правильный ответ
Ведущая причина артериальной гипертензии у лиц пожилого и старческого возраста
1) атеросклероз
2) болезнь Иценко-Кушинга
3) хронический пиелонефрит
4) феохромоцитома
! 1
№ 12
* 1 -один правильный ответ
Язвенный дефект у лиц пожилого и старческого возраста чаще локализуется в
1) желудке
2) 12-ти перстной кишке
3) слепой кишке
4) ободочной кишке
! 1
№ 13
* 1 -один правильный ответ
В возникновении язвенной болезни в пожилом возрасте наибольшее значение имеет
1) генетическая предрасположенность
2) нарушение микроциркуляции в стенке желудка
3) повышение кислотности желудочного сока
4) психоэмоциональные перегрузки
! 2
№ 14
* 1 -один правильный ответ
Задержка мочеиспускания у лиц пожилого и старческого возраста часто связана с
1) острым гломерулонефритом
2) острым пиелонефритом
3) острым циститом
4) аденомой предстательной железы
! 4
№ 15
* 1 -один правильный ответ
Причина снижения эффективности пероральных препаратов железа
1) увеличение всасывания
2) уменьшение всасывания
3) ускоренная эвакуация
4) повышение кислотности желудочного сока
! 2
№ 16
* 1 -один правильный ответ
Заболевание, встречающееся преимущественно у лиц пожилого и старческого возраста
1) гемофилия
2) болезнь Шенлейна-Геноха
3) железодефицитная анемия
4) хронический лимфолейкоз
! 4
№ 17
* 1 -один правильный ответ
Сахарный диабет у лиц пожилого и старческого возраста обусловлен
1) бактериальной инфекцией
2) психоэмоциональными перегрузками
3) абсолютным дефицитом инсулина
4) снижением чувствительности тканей к инсулину
! 4
№ 18
* 1 -один правильный ответ
Течение сахарного диабета у лиц пожилого и старческого возраста отягощают
1) атеросклероз, ожирение
2) пиелонефрит, цистит
3) бронхит, пневмония
4) гастрит, холецистит
! 1
№ 19
* 1 -один правильный ответ
Заболевание, характеризующееся повышенной ломкостью костей из-за снижения содержания в них кальция
1) артроз
2) ревматоидный артрит
3) остеохондроз
4) остеопороз
! 4
№ 20
* 1 -один правильный ответ
У лиц пожилого и старческого возраста чаще встречается
1) ревматический полиартрит
2) ревматоидный артрит
3) инфекционный артрит
4) остеохондроз позвоночника
! 4

Врожденная ломкость костей – это болезнь, обусловленная генетически, затрагивающая соединительную ткань, и, в частности, вызывающая нарушения строения и функции коллагена.
Как можно заключить по названию, связана с очень плохой структурой кости, которые становятся ломкими и хрупкими.
Несовершенный остеогенез – причины
Коллаген – это важный элемент соединительной ткани организма. В зависимости от того, в каком месте находится эта соединительная ткань и какие выполняет функции, соответствующий тип коллагена входит в её состав.
Несовершенный остеогенез – это заболевание, имеющее генетический фон, а мутация касается одного гена, ответственного за правильное строение коллагена – точнее, отвечающего за синтез альфа-цепи коллагена 1. Нарушения в структуре коллагена этого типа нарушают структуру и прочность костей, сухожилий, кожи, а также склер.

Заболевание наследуется чаще по аутосомно-доминантному принципу. Это означает, что наличие одного дефектного гена, полученного от одного из родителей, достаточно, чтобы появились симптомы болезни.
В зависимости от того, в какой степени происходит мутация, меняется степень тяжести сопутствующих болезни симптомов. Мутация может лишь незначительно снижать содержание коллагена в соединительной ткани, что отражается в легком течение болезни. Значительные нарушения, охватывающие процесс синтеза коллагена, могут быть причиной очень тяжелого состояния здоровья.
У некоторых людей болезнь передаётся по аутосомно-рецессивному принципу, это значит, что для того, чтобы дело дошло до появления симптомов, необходимо повреждение обоих генов, полученных от обоих родителей. Так может случиться, если каждый из родителей является носителем генетической мутации.
Симптомы врожденной хрупкости костей
Даже если заболевание обнаруживается у нескольких членов семьи, то ход его может существенно отличаться. Все случаи, однако, характеризуются различной степенью ослабления костной структуры, повышенной хрупкостью костей, склонностью к переломам. Повреждения могут возникнуть при ушибе, который у здорового человека ничего не вызовет, а в крайне тяжелых случаях повреждение костей возникают даже в состоянии покоя.
Среди других часто встречающихся симптомов:
- низкий рост
- деформации костей
- гипоплазия зубов
- голубые глаза
- глухота (во взрослом возрасте)
- дряблость суставов
- нестабильность суставов и связок
- частые синяки
Типы врожденной хрупкости костей
Существует несколько типов врожденной хрупкости кости. Самая мягкая форма – 1 тип, наиболее распространенный, характеризующийся низкой хрупкостью кости, дети не слишком малы и деформации костей не наблюдаются. Когда ребенок начинает делать первые шаги, появляются первые переломы, в том числе не только длинных костей, но и небольших костей рук или ног.
К сожалению, эта тенденция сопровождает ребёнка до достижения зрелости. В взрослом возрасте остеопоротические изменения в костях появляются намного раньше, чем у здоровых людей, и усиливается потеря слуха.
Тип 2 врожденной хрупкости костей называется летальной формой. Болезнь проявляется уже во время внутриутробной жизни, что приводит к ярко выраженным переломам и деформациям, часто заканчивается смертью плода. Рождение ребенка с врожденной хрупкостью костей типа 2 не даёт ему небольшие шансы на выживание в течение нескольких лет. Как правило, такие дети умирают в раннем возрасте.
Типы 3-4 – это умеренная и тяжелая форма, когда наблюдаются выраженные деформации костей, нарушения имеют разную форму и тяжесть. Они представляют собой промежуточные формы между слабой формой и летальной и могут давать разнообразную клиническую картину.
Повреждения костей могут появиться ещё до рождения ребенка. В более тяжелых случаях у ребёнка задерживается рост, деформируется осанка. Такие люди передвигаются на коляске, потому что болезнь делает невозможным нормальное функционирование. Гораздо раньше у таких людей появляется глухота. Больные требуют постоянных ортопедических консультаций.
Лечение врожденной хрупкости костей
Из-за генетического происхождения заболевания, не существует возможности полностью вылечить и внедрить эффективную терапию.
Лечение сводится к минимизации числа переломов, предотвращению больших деформаций и уменьшению боли. Применяется терапии бисфосфонатами, то есть препараты, используемыми в лечении остеопороза. Контролируют органы слуха, а также оценивают общее состояние больного, чтобы исключить проблемы в других системах организма.
Продолжительность жизни больных с врожденной хрупкостью костей зависит, в первую очередь, от типа заболевания. При типе 2 больной может не отличаться от общей популяции. При более тяжелых формах ожидаемая продолжительность жизни короче, чем у населения в целом, однако, это следует не из частых переломов, а из сопутствующих проблем с функционированием дыхательной или сердечно-сосудистой системы, что связано с деформацией грудной клетки.
Генетические заболевания обусловлены патологическими нарушениями строения генома. "Дефектный" ген может быть получен от одного из родителей и проявиться как на 100%, так и на 10%. А вот болезни с наследственной предрасположенностью значительно отличаются от генетических. Если последние излечить невозможно, то заболевания, к которым человек имеет наследственную предрасположенность, возможно нивелировать рациональным питанием, здоровым образом жизни и профилактическими мерами.
Пять генетический заболеваний позвоночника и костей
Такие болезни напрямую связанны с нарушениями генома и проявляются в виде дефектов развития скелета человека. Генетические заболевания обусловлены нерациональным формообразованием ткани или нарушениями роста. Подобные болезни носят в медицине общие название - дисплазии.
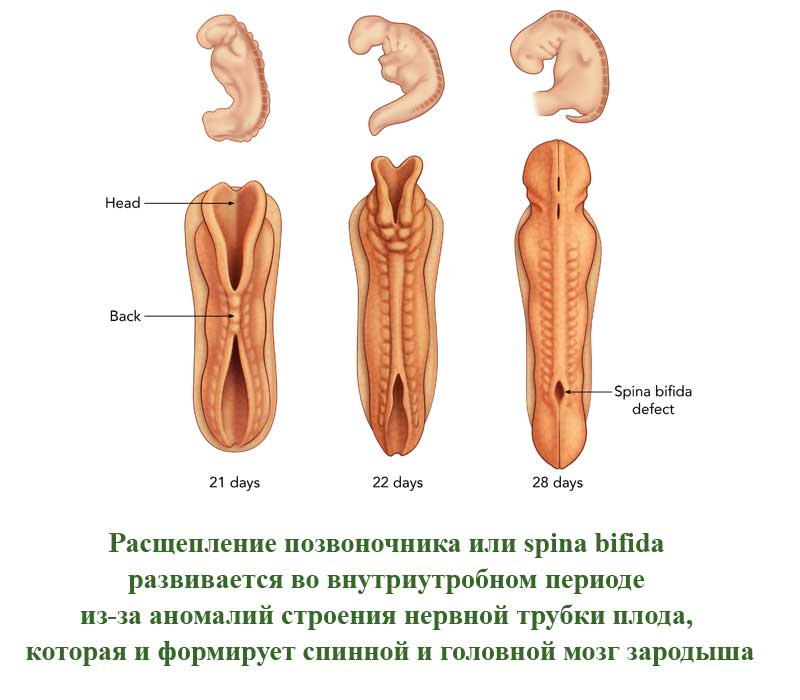
Это порок развития позвоночного столба, которое проявляется в виде недоразвитых позвонков. Такие позвонки не сомкнуты, через щель может быть виден спинной мозг. Заболевание развивается во внутриутробном периоде из-за аномалий строения нервной трубки плода, которая и формирует спинной и головной мозг зародыша. Расщепление позвоночного столба может проявляться и в закрытом виде, когда спинной мозг не виден снаружи.
В легких случаях заболевание могут обнаружить лишь при рентгеновском обследовании. А вот при самых серьезных формах болезни у ребенка могут сразу же образовываться свищи в полости позвоночника. Очень часто заболевание в тяжелых формах сопровождается параличом нижней части тела.
В более, чем 80% случаев, расщепление позвоночника сопровождается гидроцефалией спинного мозга и пороками развития головного мозга, а также - черепа.
По американской статистике, заболевание встречается у одного пациента из 1500. Российская статистика приводит следующие данные - 3 случая на 10000 человек. Однако, многие случаи расщепления позвоночника на территории СНГ остаются нераспознанными у новорожденных из-за легкой формы болезни.
Болезнь часто именуют остеопетрозом. Может протекать в двух формах:
- замедленной;
- злокачественной.
Генетическое заболевание встречается с частотой в 1 случай на 20000 пациентов. Для остеопетроза характерны такие симптомы:
- повышенная ломкость костей;
- увеличение плотности костной ткани;
- уменьшение размеров костномозговых лакун;
- нарушение гемопоэза;
- уменьшение массы костного мозга.
Генерализированный остеоклероз проявляется в достаточно раннем возрасте в виде разных беспорядочных слоев клеток костной ткани, увеличения общей массы костей и замедленном росте скелета.
При злокачественном течении болезни часто возникают внезапные переломы костей, развивается геморрагичекий синдром, жировая дистрофия органов, нарушается дентиногеез. Характерен очень небольшой рост.
В случае замедленного остеопетреза болезнь может быть выявлена лишь в 50% и протекать абсолютно бессимптомно. Выявляют заболевание случайно во время рентгена. В некоторых случаях может наблюдаться симптоматика синдрома "Кость внутри кости".
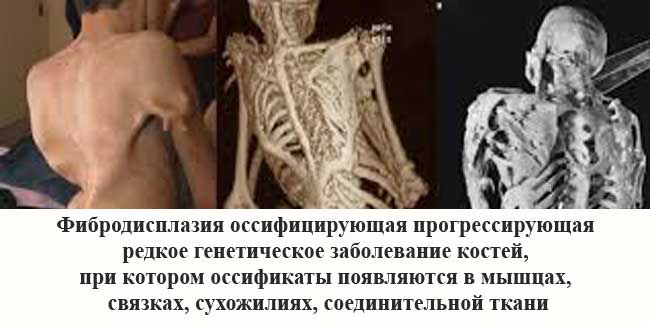
ФОП - это генетическое и очень редкое заболевание костей. При такой болезни организм начинает формировать новую костную массу в виде оссификатов в ненадлежащих местах тела, а именно внутри:
- соединительных тканей;
- связок;
- мышц;
- сухожилий.
К образованию оссификатов в организме может привести абсолютно любая травма: порез, операция, ушиб, внутримышечная инъекция или перелом. Поэтому образования такого типа удалять нельзя - на их месте костная ткань разрастется еще больше. По физиологическим признакам оссификаты совершенно не отличаются от здоровых костей.
Проблема лишь в неправильном расположении образования костной ткани. Возникает ФОП из-за мутаций гена ACVR1/ALK2. Данный ген кодирует рецептов костного морфогенетического белка. Носителем гена быть невозможно, его наличие в теле всегда вызывает развитие фибродисплазии оссифицирующей. Передается заболевание по наследству и на данный момент является неизлечимым.
Такие заболевания характеризуются чрезмерным развитием костной массы. Носят общее название - остеохондродисплазии. Гиперостозы возникают из-за генетических нарушений и патологий остеобластов и остеокластов. Наиболее часто встречаются такие формы остеохондродисплазий:
- Болезнь Лери или мелореостоз;
- пикнодизостоз.
Мелореостоз чаще всего поражает мужчин, может развиться в любом возрасте. Характеризуется болезнь избыточным образованием эндостальной или периостальной кости. Процесс может происходит в двух зонах одновременно. Зарождается болезнь Лери с поражения нижних конечностей. Процесс может переходить на все суставы, отдельные кости таза, позвоночный столб, ребра и даже череп. Все пораженные кости довольно слабо изменены и деформированы, кортикальный стой утолщен, а костномозговая полость сужена неравномерно.
Мелореостоз может протекать совершенно бессимптомно продолжительное время, однако, при значительном уменьшении габаритов костномозговых лакун развивается болевой синдром в пораженной конечности. Нога при этом может укорачиваться или увеличиваться, развивается анкилоз сустав, нарушается гемопоэз.
Пикнодизостоз проявляется в виде карликовости и остеоскрероза. В основе заболевания лежит неравномерное, чрезмерное и очаговое периостальное развитие компактной кости. Развивается явная деформация скелета в виде:
- сколиоза;
- кифоза;
- гипоплазии ключиц;
- укорочении пальцевых фаланг;
- уменьшении длины предплечий.
В молочных зубах ребенка быстро развивается кариес, склеры глаз приобретают характерных болезни голубой оттенок. На продолжительности жизни пикнодизостоз не сказывается.
Это наследственная болезнь, которая проявляется остеопенией и повышенной ломкостью костей. Несовершенный остеогенез часто носит семейный характер, при нем наблюдаются голубые склеры, аномалия развития зубов (несовершенный дентиногенез) и прогрессирующая тугоухость.

Тяжелые формы болезни приводят к гибели плода или новорожденного. Легкие и среднетяжелые формы могут протекать по-разному. При рождении отклонений от нормы может не быть, болезнь проявляется позднее и прогрессирует с возрастом.
У многих больных в грудном и детском возрасте наблюдаются множественные переломы, в пубертатном периоде переломы происходят реже, а потом снова учащаются. У женщин риск переломов особенно велик во время беременности и в постменопаузе.
У некоторых женщин с легким течением болезни переломы развиваются только в постменопаузе, и в этом случае несовершенный остеогенез трудно отличить от постклимактерического остеопороза. Классификация. Чаще всего применяют классификацию, разработанную Силленсом.
Классификация несовершенного остеогенеза
Тип I: Незначительная хрупкость костей, окраска склер: голубые, изменения зубов: только при типе IB, тугоухость: у большинства больных, тип наследования: А-Д
Тип II: Чрезвычайная хрупкость костей, окраска склер: голубые, изменения зубов: иногда, тугоухость: нет данных, тип наследования: С, реже А-Р
Тип III: Выраженная хрупкость костей, окраска склер: голубоватые при рождении, изменения зубов: иногда, тугоухость: часто, тип наследования: А-P или А-Д
Тип IV: Разной степени хрупкость костей, окраска склер: голубые, изменения зубов: только при типе IVB, тугоухость: часто, тип наследования: А-Д
Несовершенный остеогенез типа I протекает легче всего и наследуется аутосомно-рецессивно. Большинство больных имеют отчетливо голубые склеры.
Несовершенный остеогенез типа I разделяют на подтипы А и В (последний отличается наличием несовершенного дентиногенеза).
При несовершенном остеогенезе типа II происходит гибель плода или новорожденного. На основании особенностей изменения скелета, выявляемых рентгенологически, несовершенный остеогенез типа II разделяют на 3 подтипа (А, В и С).
Несовершенный остеогенез типов III и IV протекает легче, чем типа II; от типа I отличается тем, что склеры обычно нормального цвета и лишь в трудном возрасте могут быть голубоватыми. Несовершенный остеогенез типа III (в отличие от типа IV) прогрессирует с возрастом.
Кроме того, тип III наследуется аугосомно-доминантно или аутосомно-рецессивно, а тип IV—только аутосомно-доминантно.
Течение может быть различным, тип наследования не всегда легко установить, так как болезнь часто вызывается новой мутацией, и многие родители после рождения ребенка с тяжелой формой несовершенного остеогенеза остерегаются заводить других детей. По элям и другим причинам выделять тип IV практически не имеет смысла. Достаточно разделять несовершенный остеогенезна легкий (тип I), смертельный (тип II) и среднетяжелый (тип III).
Распространенность несовершенного остеогенеза типа I составляет 1 на 30 000, типа II — примерно 1 на 60 000 новорожденных, а распространенность трех тяжелых форм, распознаваемых при рождении (типы И, III и ГУ), достигает 1 на 20 000.
При несовершенном остеогенезе типа I хрупкость костей может приводить к ограничению физической активности, а может и не причинять больному никаких неудобств. Свод черепа на рентгенограммах выглядит пятнистым из-за небольших участков нарушенного окостенения.
При несовершенном остеогенезе типа II кости и другие соединительные ткани такие непрочные, что это приводит к тяжелым травмам плода во время беременности и родов (рис. 348.5). Минерализация многих костей не завершена, характерны множественные утолщения и переломы ребер, деформация длинных трубчатых костей.
По неясным причинам длинные трубчатые кости могут утолщаться или, наоборот, истончаться. При несовершенном остеогенезе типов III и IV множественные переломы при незначительных травмах вызывают грубую деформацию костей.
Кифосколиоз приводит к дыхательной недостаточности, частым пневмониям и формированию легочного сердца. Неблагоприятный признак — появление участков обызвествления на концах длинных трубчатых костей, на рентгенограмме напоминающих воздушную кукурузу. Платибазия и сообщающаяся гидроцефалия могут привести к прогрессирующим неврологическим расстройствам.
Для всех форм несовершенного остеогенеза характерна остеопения. Степень остеопении бывает трудно оценить, так как повторные переломы приводят к ограничению физической активности и еще большему снижению плотности кости. Но, как это ни странно, переломы срастаются довольно хорошо.
Склеры могут быть белыми, слегка голубоватыми или ярко-голубыми. Голубой цвет дает сосудистая оболочка глаза, просвечивая через более тонкий, чем обычно, слой коллагена склеры. В некоторых семьях голубые склеры передаются по наследству, но не сопровождаются ломкостью костей.
Поражение зубов наблюдается не всегда; оно может быть значительным или проявляться умеренным изменением окраски. Эмаль, как правило, не изменена, характерный янтарный, желтовато-коричневый или серо-голубой цвет зубов связан с недостаточностью дентина. Молочные зубы обычно мелкие, а постоянные зубы могут иметь форму колокола (сужаются к основанию). У некоторых больных зубы легко ломаются, и их приходится удалять.
Изменения дентина связаны с тем, что в нем много коллагена I типа. Похожие изменения зубов иногда встречаются и в отсутствие несовершенного остеогенеза.
Слух обычно начинает снижаться в возрасте 10— 20 лет, а к 30 годам тугоухость развивается у 90% больных. Тугоухость может быть разной степени, кондуктивной, нейросенсорной или смешанной. Обычно наблюдаются аномалии развития среднего уха, недостаточная минерализация в одних участках и патологические очаги обызвествления — в других участках слуховых косточек.
Поражение других органов и тканей проявляется тонкостью кожи, склонностью к образованию рубцов, разболтанностью и привычными вывихами суставов, как и при синдроме Элерса—Данло. Поражение сердечнососудистой системы может проявляться пролапсом митрального клапана, митральной и аортальной недостаточностью и разрывами крупных кровеносных сосудов.
У некоторых больных по неизвестным причинам повышены концентрация Т4 в сыворотке и основной обмен, что проявляется гипертермией и повышенным потоотделением.
Как уже говорилось, в большинстве случаев несовершенного остеогенеза обнаруживают мутации одного из двух генов, кодирующих коллаген I типа.
По меньшей мере у трети больных вследствие неизвестной мутации снижена концентрация мРНК про-а1(1)-цепей, которых при этом синтезируется меньше, чем про-α2(1)-цепей. При тяжелых формах несовершенного остеогенеза (типы II, III и IV) эффект мутаций многократно усиливается в результате действия трех механизмов, описанных выше.
При мутациях, изменяющих структуру белка в месте его расщепления проколлаген-М-эндопептидазой, происходит накопление коллагена с сохраненным N-концевым пропептидом, что приводит к разболтанности суставов, как при синдроме Элерса—Данло типа VII.
Мутации, изменяющие структуру средней или С-концевой частей молекулы, вызывают тяжелые и часто смертельные формы несовершенного остеогенеза. Однако найти соответствие между той или иной мутацией и определенной клинической картиной довольно трудно. Изредка встречаются гомозиготы по мутантным аллелям, кодирующим про-α1- или про-α2-цепи.
Большинство смертельных форм несовершенного остеогенеза — это результат новых мутаций с аутосомно-доминантным типом наследования. Тем не менее вероятность рождения второго ребенка со смертельной формой несовершенного остеогенеза в той же семье составляет около 7% из-за гонадного мозаицизма у одного из родителей.
Обнаружено, что у некоторых мужчин, чьи дети страдают несовершенным остеогенезом типа II, часть сперматозоидов несет мутантный аллель. Кроме того, у здоровых родителей таких детей можно выявить мозаицизм соматических клеток, таких, как фибробласты, лейкоциты и клетки корней волос. Возможность гонадного мозаицизма следует учитывать при медико-генетическом консультировании здоровых родителей, имеющих ребенка с тяжелой формой несовершенного остеогенеза.
Основанием для постановки диагноза обычно служит сочетание клинической картины (множественные переломы, голубые склеры, несовершенный дентиногенез) и семейного анамнеза. Следует исключить другие причины патологических переломов: жестокое обращение с ребенком, истощение, авитаминозы, злокачественные новообразования, а также другие наследственные болезни, например хондродисплазии или гипофосфатазию.
При рентгенологическом исследовании обнаруживают разрежение кости, что можно подтвердить с помощью денситометрии; вопрос о необходимости биопсии кости спорен. Более чем у половины больных имеется дефект проколлагена I типа.
Путем инкубации культуры фибробластов с мечеными аминокислотами и последующего электрофореза проколлагена в полиакриламидном геле можно выявить следующие нарушения, препятствующие сборке тройных спиралей:
1) снижение скорости синтеза про-а1(1)-цепей по сравнению с про-а2(1)-цепями;
2) укорочение или удлинение про-а(1)-цепей;
3) изменение структуры про-а(1)-цепей в результате замен аминокислот и нарушения посттрансляционной модификации. Сами мутации выявляют путем анализа геномной ДНК или кДНК.
Поскольку каждая семья обычно имеет свою собственную мутацию, для ее выявления необходимо определить последовательность не менее 5000 оснований в каждом из двух генов. Если мутацию удается установить, становятся возможными пренатальная диагностика и выявление носителей мутации среди членов семьи больного с помощью ПЦР.
| Возраст | Диагноз | Отличительные признаки |
| Новорожденные | Г ипофосфатазия |
Нарушение остеогенеза при неподвижности Цинга
Опухоли коры надпочечников
Лечение
Несовершенный остеогенез не поддается лечению, однако, несмотря на тяжелые деформации костей, многие больные успешно работают. При легких формах болезни после полового созревания риск переломов снижается, и болезнь практически не требует лечения.
Исключение составляют женщины во время беременности и в постменопаузе, когда переломы учащаются. Программа помощи тяжелобольным детям должна включать ЛФК, хирургическое лечение переломов и деформаций костей и специальную профессиональную подготовку.
Многие переломы сопровождаются лишь небольшим смещением и отеком мягких тканей, и для их лечения бывает достаточно иммобилизации или небольшого вытяжения в течение 1—2 нед с последующим наложением легкой полимерной шины. Если нет сильных болей, можно рано начать ЛФК. Физическая нагрузка в разумном объеме позволяет предотвратить гипокинетический остеопороз.
Некоторые врачи рекомендуют исправлять деформацию конечностей путем введения стальных стержней в длинные трубчатые кости, но этот метод спорен и достаточно дорог. Лечение пневмонии и легочного сердца проводят обычными методами. При выраженной тугоухости показаны стапедэктомия и протезирование стремечка.
Больных со среднетяжелыми и тяжелыми формами болезни должен регулярно осматривать невропатолог. Примерно у половины детей удается стимулировать рост путем введения СТГ. Обсуждается вопрос о лечении остеопении дифосфонатами, но контролируемых исследований пока не проводилось.
Довольно полезными оказались ортопедическая программа Блека и комплексная программа Марини. Для больных и их родителей очень важна психологическая поддержка; в некоторых странах для этого созданы специальные общественные организации. УЗИ позволяет выявить тяжелое поражение плода при сроке беременности около 16 нед, а биопсия ворсин хориона — дефект про-а-цепей или непосредственно мутацию гена при сроке 8—12 нед.

Несовершенный остеогенез – врожденное расстройство, характеризующееся хрупкостью костей, которые имеют тенденцию к повышенной ломкости. Люди с несовершенным остеогенезом рождаются с дефектами соединительной ткани или дефицитом коллагена типа I. В большинстве случаев расстройство вызвано мутациями генов COL1A1 и COL1A2. Заболевание встречается у одного из 20000 новорожденных.
Типы несовершенного остеогенеза
Различают восемь типов несовершенного остеогенеза.
Тип I является наиболее распространенным, отличается от остальных тем, что коллаген обладает нормальными качественными свойствами, однако вырабатывается в недостаточном количестве. Симптомами несовершенного остеогенеза типа I являются:
- Ломкость костей;
- Слабость суставов;
- Слегка выступающие глаза;
- Пониженный мышечный тонус;
- Ранняя потеря слуха у некоторых детей;
- Незначительное искривление позвоночника;
- Обесцвечивание склер (белков глаз), что, как правило, придает им сине-карий оттенок.
Симптомами несовершенного остеогенеза типа II являются:
- Недостаточное содержание коллагена;
- Дыхательные проблемы из-за слаборазвитых легких;
- Невысокий рост;
- Деформация костей.
II тип может быть подразделен на группы A, B, C, которые различаются благодаря радиографическому исследованию длинной трубчатой кости и ребер.
В большинстве случаев больные умирают в течение первого года жизни в связи с дыхательной недостаточностью или внутричерепным кровоизлиянием.
Несовершенный остеогенез III типа характеризуется следующими симптомами:
- Коллаген вырабатывается в достаточном количестве, но не достаточного качества;
- Легкой ломкостью костей, иногда даже при рождении;
- Деформацией костей;
- Возможными проблемами с дыханием;
- Невысоким ростом, искривлением позвоночника, иногда также бочковидностью грудной клетки;
- Слабостью связочного аппарата суставов;
- Слабостью мускульного тонуса рук и ног;
- Обесцвечиванием склер (глазных белков);
- Ранней потерей волос.
Продолжительность жизни может быть нормальной, хотя и с тяжёлыми физическими недостатками.
IV тип несовершенного остеогенеза характеризуется симптомами:
- Коллаген вырабатывается в достаточном количестве, но не достаточно высокого качества;
- Кости разрушаются легко, особенно до полового созревания;
- Низким ростом, искривлением позвоночника и бочкообразной формой грудной клетки;
- Слабой или средней деформацией костей;
- Ранней потерей слуха.

V тип несовершенного остеогенеза имеет те же клинические признаки, как тип IV. Отличается внешним видом решетчатой кости, радиальной дислокацией головы и смешанной потерей слуха, приводит к кальцификации мембраны между двумя костями предплечья.
Тип VI несовершенного остеогенеза имеет те же клинические признаки, что и тип IV, но отличается уникальностью гистологических данных костной ткани. Причинами несовершенного остеогенеза типа VI являются утрата функций и мутация гена Серпин F1.
Причиной несовершенного остеогенеза типа VII является мутация протеина хрящевой ткани, а несовершенный остеогенез типа VIII является тяжелым и смертельным расстройством, которое связано с изменением белка, содержащего лейцин и пролин.
Лечение несовершенного остеогенеза
Лечения несовершенного остеогенеза не существует, так как это заболевание является врожденным (генетическим). Лечение направлено на повышение общей прочности костей, чтобы предотвратить и приостановить их дальнейшее разрушение. Также применяют терапию бисфосфонатом, которая способствует увеличению костной массы, уменьшению боли в костях и их разрушения. В тяжелых случаях применяют хирургическое вмешательство и размещают стержни внутри костей.
Видео с YouTube по теме статьи:
Информация является обобщенной и предоставляется в ознакомительных целях. При первых признаках болезни обратитесь к врачу. Самолечение опасно для здоровья!
Читайте также:
