
Системные заболевания соединительной ткани дерматомиозит
Системные заболевания соединительной ткани
Системные заболевания соединительной ткани, или диффузные заболевания соединительной ткани – группа заболеваний, характеризующихся системным типом воспаления различных органов и систем, сочетающимся с развитием аутоиммунных и иммунокомплексных процессов, а также избыточным фиброзообразованием.
Группа системных заболеваний соединительной ткани включает в себя следующие заболевания:
1) системная красная волчанка;
2) системная склеродермия;
3) диффузный фасциит;
4) дерматомиозит (полимиозит) идиопатический;
5) болезнь (синдром) Шегрена;
6) смешанное заболевание соединительной ткани (синдром Шарпа);
7) ревматическая полимиалгия;
8) рецидивирующий полихондрит;
9) рецидивирующий панникулит (болезнь Вебера – Крисчена).
Кроме того, в настоящее время к этой группе относят болезнь Бехчета, первичный антифосфолипидный синдром, а также системные васкулиты.
Системные заболевания соединительной ткани объединены между собой основным субстратом – соединительной тканью – и схожим патогенезом.
В настоящее время доказано, что при системных заболеваниях соединительной ткани происходят глубокие нарушения иммунного гомеостаза, выражающиеся в развитии аутоиммунных процессов, то есть реакций иммунной системы, сопровождающихся появлением антител или сенсибилизированных лимфоцитов, направленных против антигенов собственного организма (аутоантигенов).
Кроме близкого патогенеза, для всех системных заболеваний соединительной ткани являются характерными следующие черты:
1) мультифакториальный тип предрасположения с определенной ролью иммуногенетических факторов, связанных с шестой хромосомой;
2) единые морфологические изменения (дезорганизация соединительной ткани, фибриноидные изменения основного вещества соединительной ткани, генерализованное поражение сосудистого русла: васкулиты, лимфоидные и плазмоклеточные инфильтраты и др.);
3) схожесть отдельных клинических признаков, особенно в ранней стадии болезни (например, синдром Рейно);
4) системность, полиорганность поражения (суставы, кожа, мышцы, почки, серозные оболочки, сердце, легкие);
5) общие лабораторные показатели активности воспаления;
6) общие групповые и характерные для каждой болезни иммунологические маркеры;
7) близкие принципы лечения (противовоспалительные средства, иммуносупрессия, экстракорпоральные методы очищения и пульскортикостероидная терапия в кризовых ситуациях).
Этиология системных заболеваний соединительной ткани рассматривается с позиций мультифакторной концепции аутоиммунитета, согласно которой развитие этих болезней обусловлено взаимодействием инфекционных, генетических, эндокринных и внешнесредовых факторов (т. е. генетическая предрасположенность + внешнесредовые факторы, такие как стресс, инфекция, переохлаждение, инсоляция, травма, а также действие половых гормонов, в основном женских, беременность, аборты – системные заболевания соединительной ткани).
Чаще всего внешнесредовые факторы либо обостряют скрыто протекающую болезнь, либо являются, при наличии генетической предрасположенности, пусковыми моментами возникновения системных заболеваний соединительной ткани. До сих пор ведутся поиски конкретных инфекционных этиологических факторов, в первую очередь вирусных. Возможно, что происходит еще внутриутробное инфицирование, об этом свидетельствуют эксперименты на мышах.
В настоящее время накоплены косвенные данные о возможной роли хронической вирусной инфекции. Изучается роль пикорнавирусов при полимиозите, РНК-содержащих вирусов – при кори, краснухе, парагриппе, паротите, системной красной волчанке, а также ДНК-содержащих герпетических вирусов – Эпштейна – Барр цитомегаловируса, вируса простого герпеса.
Хронизация вирусной инфекции связана с определенными генетическими особенностями организма, что позволяет говорить о нередком семейно-генетическом характере системных заболеваний соединительной ткани. В семьях больных, по сравнению с семьями здоровых и с популяцией в целом, чаще наблюдаются различные системные заболевания соединительной ткани, особенно среди родственников первой степени родства (сестер и братьев), а также более частое поражение монозиготных близнецов, чем дизиготных.
Многочисленными исследованиями показана ассоциация между носительством определенных HLA-антигенов (которые размещаются на коротком плече шестой хромосомы) и развитием конкретного системного заболевания соединительной ткани.
Для развития системных заболеваний соединительной ткани наибольшее значение имеет носительство II класса HLA-D-генов, локализующихся на поверхности В-лимфоцитов, эпителиальных клеток, клеток костного мозга и др. Например, системная красная волчанка ассоциируется с DR3-антигеном гистосовместимости. При системной склеродермии отмечается накопление Al, В8, DR3-антигенов в сочетании с DR5-антигеном, а при первичном синдроме Шегрена – высокая связь с HLA-B8 и DR3.
Таким образом, механизм развития таких сложных и многоликих заболеваний, какими являются системные заболевания соединительной ткани, до конца не изучен. Однако практическое применение диагностических иммунологических маркеров заболевания и определение его активности позволит улучшить прогноз при этих заболеваниях.
Остановимся подробнее на некоторых наиболее значимых системных заболеваниях соединительной ткани.
Системная красная волчанка
Системная красная волчанка – это хроническое прогрессирующее полисиндромное заболевание преимущественно молодых женщин и девушек (соотношение больных женщин и мужчин 10: 1), которое развивается на фоне генетически обусловленного несовершенства иммунорегуляторных механизмов и ведет к неконтролируемому синтезу антител к собственным тканям организма с развитием аутоиммунного и иммунокомплексного хронического воспаления (В. А. Насонова, 1989 г.).
По своей сущности системная красная волчанка является хроническим системным аутоиммунным заболеванием соединительной ткани и сосудов, характеризующимся множественными поражениями различной локализации: кожи, суставов, сердца, почек, крови, легких, центральной нервной системы и других органов. При этом висцеральные поражения определяют течение и прогноз заболевания.
Распространенность системной красной волчанки увеличилась за последние годы с 17 до 48 человек на 100 тыс. населения. Вместе с тем улучшение диагностики, раннее распознавание доброкачественных вариантов течения со своевременным назначением адекватного лечения привели к удлинению продолжительности жизни больных и улучшению прогноза в целом.
Начало заболевания часто можно связать с длительным пребыванием на солнце в летний период, перепадами температуры при купании, введением сывороток, приемом некоторых лекарственных средств (в частности, периферических вазодилататоров из группы гидролазинов), стрессами, а также системная красная волчанка может начаться после родов, перенесенного аборта.
Подострое течение. Начало как бы исподволь, с общих симптомов, артралгий, рецидивирующих артритов, разнообразных неспецифических поражений кожи в виде дисковидной волчанки, фотодерматозов на лбу, шее, губах, ушах, верхней части груди. Отчетлива волнообразность течения. Развернутая картина болезни формируется через 2–3 года.
1) поражение сердца, чаще в виде бородавчатого эндокардита Либмана – Сакса с отложениями на митральном клапане;
2) часты миалгии, миозиты с атрофией мышц;
3) всегда присутствует синдром Рейно, довольно часто заканчивающийся ишемическим некрозом кончиков пальцев;
6) нефрит, который не достигает такой степени активности, как при остром течении;
7) радикулиты, невриты, плекситы;
8) упорные головные боли, утомляемость;
9) анемия, лейкопения, тромбоцитопения, гипергаммаглобулинемия.
Хроническое течение. Заболевание длительное время проявляется рецидивами различных синдромов: полиартрита, реже – полисерозита, синдромом дискоидной волчанки, синдромами Рейно, Верльгофа, эпилептиформным. На 5—10-м году болезни присоединяются другие органные поражения (преходящий очаговый нефрит, пневмонит).
В качестве начальных признаков заболевания следует отметить кожные изменения, лихорадку, исхудание, синдром Рейно, диарею. Больные жалуются на нервозность, плохой аппетит. Обычно, за исключением хронических олигосимптомных форм, заболевание прогрессирует достаточно быстро и развивается полная картина болезни.
При развернутой картине на фоне полисиндромности весьма часто начинает доминировать один из синдромов, что позволяет говорить о люпус-нефрите (наиболее часто встречающаяся форма), люпус-эндокардите, люпус-гепатите, люпус-пневмоните, нейролюпусе.
К поражениям кожных покровов относят капилляриты – мелкоотечную геморрагическую сыпь на подушечках пальцев рук, ногтевых ложах, ладонях. Встречается поражение слизистой оболочки твердого нёба, щек и губ в виде энантемы, иногда с изъязвлениями, стоматитом.
Довольно рано наблюдается выпадение волос, увеличивается ломкость волос, поэтому на этот признак следует обращать внимание.
Поражение серозных оболочек отмечается у подавляющего числа больных (90 %) в виде полисерозита. Наиболее часто встречаются плеврит и перикардит, реже – асцит. Выпоты необильные, с тенденцией к пролиферативным процессам, ведущим к облитерации плевральных полостей и перикарда. Поражение серозных оболочек кратковременно и обычно диагностируется ретроспективно по плевроперикардиальным спайкам или утолщению костальной, междолевой, медиастинальной плевры при рентгенологическом исследовании.
Поражение опорно-двигательного аппарата проявляет себя полиартритом, напоминающим ревматоидный артрит. Это наиболее часто встречающийся признак системной красной волчанки (у 80–90 % больных). Характерно преимущественно симметричное поражение мелких суставов кистей, лучезапястных, а также голеностопных суставов. При развернутой картине болезни определяется дефигурация суставов за счет периартикулярного отека, а в последующем – развитие деформаций мелких суставов. Суставной синдром (артрит или артралгии) сопровождается диффузными миалгиями, иногда тендовагинитами, бурситами.
Поражение сердечно-сосудистой системы встречается достаточно часто, примерно у трети больных. На различных этапах заболевания выявляют перикардиты с тенденцией к рецидивам и облитерации перикарда. Наиболее тяжелой формой поражения сердца является бородавчатый эндокардит Лимбана – Сакса с развитием вальвулитов митрального, аортального и трехстворчатого клапанов. При длительном течении процесса можно выявить признаки недостаточности соответствующего клапана. При системной красной волчанке достаточно часто встречается миокардит очагового (практически никогда не распознается) или диффузного характера.
В. А. Насонова обращает внимание на то, что поражения сердечно-сосудистой системы при системной красной волчанке имеют место чаще, чем это обычно удается распознать. Вследствие этого следует уделять внимание жалобам больных на боли в области сердца, сердцебиения, одышку и т. д. Больные системной красной волчанкой нуждаются в тщательном кардиологическом обследовании.
Поражение сосудов может проявляться в виде синдрома Рейно – расстройстве кровоснабжения кистей и (или) стоп, усиливающемся под воздействием холода или волнения, характеризующемся парестезиями, бледностью и (или) цианотичностью кожи II–V пальцев, их похолоданием.
Поражение легких. При системной красной волчанке наблюдаются изменения двоякого характера, как вследствие вторичной инфекции на фоне пониженной физиологической иммунологической реактивности организма, так и волчаночного васкулита легочных сосудов – люпус-пневмонита. Возможно также осложнение, возникающее как следствие люпус-пневмонита – вторичная банальная инфекция.
Если диагностика бактериальной пневмонии не представляет затруднений, то диагностика люпус-пневмонита иногда затруднительна вследствие ее мелкоочаговости с преимущественной локализацией в интерстиции. Люпус-пневмонит протекает либо остро, либо тянется месяцами; характеризуется малопродуктивным кашлем, нарастающей одышкой при скудных аускультативных данных и типичной рентгенологической картиной – сетчатым строением легочного рисунка и дисковидными ателектазами, преимущественно в средне-нижних долях легкого.
Поражение почек (волчаночный гломерулонефрит, люпус-нефрит). Часто является определяющим в исходе заболевания. Обычно он характерен для периода генерализации системной красной волчанки, однако иногда является и ранним признаком болезни. Варианты поражения почек различны. Очаговый нефрит, диффузный гломерулонефрит, нефротический синдром. Поэтому изменения характеризуются в зависимости от варианта либо скудным мочевым синдромом (протеинурией, цилиндрурией, гематурией), либо – чаще – отечно-гипертензивной формой с хронической почечной недостаточностью.
Поражение желудочно-кишечного тракта проявляется в основном субъективными признаками. При функциональном исследовании можно иногда обнаружить неопределенную болезненность в эпигастрии и в области проекции поджелудочной железы, а также признаки стоматита. В ряде случаев развивается гепатит – при обследовании отмечают увеличение печени, ее болезненность.
Поражение центральной и периферической нервной системы описывается всеми авторами, изучавшими системную красную волчанку. Характерно разнообразие синдромов: астено-вегетативный синдром, менингоэнцефалит, менингоэнцефаломиелит, полиневрит-радикулит. Поражения нервной системы возникают в основном за счет васкулитов. Иногда развиваются психозы – либо на фоне кортикостероидной терапии как ее осложнение, либо по причине ощущения безвыходности страдания. Может быть эпилептический синдром.
Синдром Верльгофа (аутоиммунная тромбоцитопения) проявляется высыпаниями в виде геморрагических пятен различной величины на коже конечностей, груди, живота, на слизистых оболочках, а также кровотечениями после незначительных травм.
Если определение варианта течения системной красной волчанки важно для оценки прогноза заболевания, то для определения тактики ведения больного необходимо уточнение степени активности патологического процесса.
Основные задачи комплексной патогенетической терапии:
1) подавление иммунного воспаления и иммунокомплексной патологии;
2) предупреждение осложнений иммуносупрессивной терапии;
3) лечение осложнений, возникающих в процессе проведения иммуносупрессивной терапии;
4) воздействие на отдельные, резко выраженные синдромы;
5) удаление из организма циркулирующих иммунных комплексов и антител.
Основным методом лечения системной красной волчанки является кортикостероидная терапия, которая остается средством выбора даже в начальных стадиях болезни и при минимальной активности процесса. Поэтому больные должны находиться на диспансерном учете с тем, чтобы при первых же признаках обострения заболевания врач своевременно мог назначить кортикостероиды. Доза глюкокортикостероидов зависит от степени активности патологического процесса.
При III степени активности – лечение в условиях специализированного или терапевтического стационара – пульстерапия глюкокортикостероидами, иммунодепрессанты. Пульстерапия: метипред – 1000 мг 3 дня подряд внутривенно капельно, одновременно глюкокортикостероиды per os – 40–60 мг в сутки до получения эффекта (снижение активности волчаночного процесса). Параллельно целесообразно делать плазмаферез 2–3—4 процедуры (с целью удаления ЦИК). В некоторых случаях можно проводить гемосорбцию.
Если нельзя использовать глюкокортикостероиды (непереносимость, резистентность), назначаются депрессанты в таблетках (метотрексат – 7,5 мг в неделю) или пульс-терапия: 20 мг/кг массы тела циклофосфамида 1 раз в месяц в течение 6 месяцев внутривенно с последующим плазмаферезом.
При уменьшении степени активности заболевания дозы глюкокортикостероидов снижают по 10 мг в неделю до 20 мг, а затем по 2,5 мг в месяц до поддерживающей дозы 5—10 мг в сутки. Никогда не отменять глюкокортикостероиды летом!
При II степени активности патологического процесса подавляющая доза преднизолона составляет 30–40 мг в сутки, а при I степени активности – 15–20 мг в сутки. Если через 24–48 ч состояние больного не улучшается, то первоначальную дозу увеличивают на 25–30 %, и если эффект наблюдается, то дозу оставляют без изменений. После достижения клиниколабораторного эффекта (снижение активности процесса), что обычно бывает после 2 месяцев кортикостероидной терапии, а при нефротическом синдроме или признаках поражения почек – спустя 3–6 месяцев, дозу преднизолона постепенно снижают до поддерживающей (5—10 мг), которую принимают годами.
Данный текст является ознакомительным фрагментом.
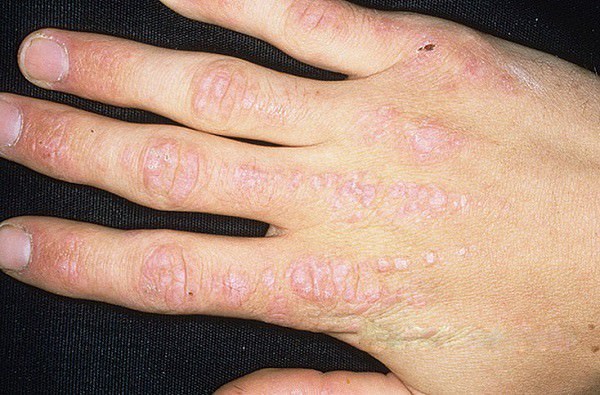
Среди заболеваний соединительных тканей есть грозная патология — системный дерматомиозит, которую называют также болезнью Вагнера или лиловой болезнью (последнее название обусловлено характерным лиловым цветом кожных пятен и эритем). Опасность ее в поражении не только кожи, но и мышц (как скелетных, так и гладких), внутренних органов (сердца, легких, ЖКТ, эндокринных желез), сосудов и нервов. Рассмотрим дерматомиозит, его симптомы и лечение, но вначале фото: Кисть руки больного дерматомиозитом.
Что такое дерматомиозит (ДМ)
Дерматомиозит — редкое воспалительное заболевание мягких тканей с кожными проявлениями.
ДМ болеют несколько человек из 100 000, причем взрослые заболевают в два раза чаще, чем дети, а подверженность женщин этой болезни несколько выше. Более точные цифры: дети — около трех чел. из 100 тыс.; мужчины — около 6 из 100 тыс.
Замечено большее распространение дерматомиозита в южных и южно-европейских странах весной и летом, из-за чего сделано предположение о возможном влиянии солнечных лучей на развитие патологии. Также эмпирические наблюдения помогли установить связь между инфекционными заболеваниями, прививками и некоторыми лекарствами и развивающимся через какое-то время (скрытый период может достигать трех месяцев) дерматомиозите.
Было установлено, что развитию ДМ способствуют:
- из вирусных заболеваний — грипп, парвовирус, гепатит В и А, ящур, полиомиелит, ринит;
- бактериальных — боррелиоз, гемолитический стрептококк;
- вакцинации — прививки от кори, свинки, тифа, холеры;
- лекарственные препараты — пеницилламин, СТГ (соматотропный гормон).
Дерматомиозит классифицируют по типу и характеру течения.
- идиопатический (причина первичной патологии неизвестна);
- паранеопластический (вызван онкологическим процессом);
- ювенильный или детский (наблюдается преимущественно в юности и детстве);
- полимиозит (без кожных проявлений, но с симптомами диффузного поражения многих тканей);
- дерматополимиозит (присутствуют как кожные поражения, так и других системных тканей).
По характеру течения:
- острый (с быстрым нарастанием симптомов);
- подострый (умеренная симптоматика);
- асимптомный (без явных клинических признаков, диагноз устанавливается на основании рентгенологического и функционального диагностирования);
- хронический (медленное развитие болезни).
ДМ обычно начинается с кожного поражения. Симптомы мышечных патологий могут присоединиться через несколько месяцев, а то и лет. Одновременно развитие миозита в коже и мышцах — нонсенс, то есть исключительно редкое явление.
Главные признаки ДМ — это сыпь различных типов.
- Симптом Готтрона — узелки или бляшки, порой с симптомами шелушения, или большие пятна красного или розового цвета, локализованные на сгибательных суставных поверхностях, преимущественно кистей рук (межфаланговых или пястнофаланговых), а также локтевых и коленных суставов.
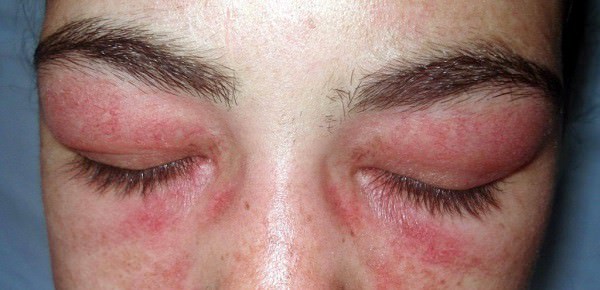
- Гелиотропная сыпь лилового цвета по типу очков около глаз, под бровями, или мантии на верхней области конечностей, спины и ключицы. Кожные высыпания возможны также на животе, ягодицах, бедрах и голени. Также бывают темные бордово-синие разветвляющиеся эритемы.
- Начальный симптом дерматомиозита — это покраснение и припухание кожи у границ ногтей.
- При дерматомиозите, отягощенными кальцинозом, появляются гнойные раны, наполненные крошкообразной массой — признак отложения кальциевых депозитов под кожей. Кальциноз наблюдается в пять раз чаще при ювенильном ДМ.
Полимиозит способен чаще всего поражать мышцы скелета, суставы, легкие, сердце, эндокринную систему.
В соединительных тканях начинаются структурные преобразования, они уплотняются, разрастаются, а затем к этому процессу может присоединиться кальциноз. Образование отложений солей кальция может происходить достаточно глубоко и не проявляться внешне, но кальциноз всегда дает типичную картину одиночных или множественных узлов на рентгене.
- При поражении скелетных мышц, которое происходит в основном симметрично, у больного развивается мышечная атрофия, возможна фибротизация мышц и образование контрактур.
- Страдают чаще плечевые, тазобедренные и брюшные мышцы (в основном пресса), а также шейные сгибатели. Это проявляется в трудностях при определенных движениях, требующих усилий, таких как подъем вверх по лестнице или попытке приподняться с сидения, особенно с низкого.
- Наиболее неблагоприятна форма мышечного полимиозита с поражением дыхательных мышц. При этом, кроме угрозы дыхательной недостаточности, у больного изменяется голос (появляется хрипота или гнусавость).
Легочная форма может быть осложнением полимиозита и считается наиболее опасной.
Привести к ней могут:
- поражение полимиозитом дыхательных мышц и м — ц диафрагмы, приведшее к недостаточной вентиляции легких;
- вирусные и бак. инфекции;
- попадание воды или пищи в легкие из пищевода;
- интерстициальная пневмония;
- злокачественные опухоли и метастазы в легких.
Симптомы легочного полимиозита:
- поверхностность и учащенность дыхания;
- прогрессирующая одышка;
- гипоксемия (понижение О2 в крови);
- шумы в легких;
- фиброз легочной ткани и альвеол.
Острый легочный миозит имеет неблагоприятный прогноз: при нем очень быстро нарастает дыхательная недостаточность, а до 10% случаев заканчиваются фиброзом легких, что для легких означает ограниченную возможность осуществления дыхательной функции.
Асимптомная форма свойственна для ранних стадий паранеопластического ПМ. При развитии злокачественного процесса появляются далее симптомы:
- инспираторной одышки;
- надсадного сухого кашля;
- в нижних отделах прослушиваются шумы и хрипы (крепитация).
Суставный ПМ чаще всего развивается из-за инфекций и поражает периартикулярные соединительные ткани (суставные мышцы, связки, сухожилия, капсулы). Он характеризуется болями, тугоподвижностью суставов, особенно в утреннее время. Порой происходит выпот в сустав, из-за чего он может опухать.
Эта форма ПМ обратима и имеет благоприятный прогноз: после проведенной антибактериальной/антивирусной терапии, приема цитостатиков суставные проявления регрессируют.
Обычно это заболевание проявляется в поражении миокарда, но могут пострадать все три сердечные оболочки, а также коронарные артерии.
Симптомы при сердечном ПМ:
- учащение сердечного ритма;
- аритмия;
- приглушённость тонов сердца;
- при неблагоприятном исходе инфаркт.
Желудочно-кишечный полимиозит вызывается поражением кровеносных и нервных сосудов, и гладкой мускулатуры ЖКТ.
В результате этого нарушается питание слизистой оболочки и перистальтика кишечника:
- начинают проявляться симптомы гастрита и колита;
- на внутренних стенках желудка и кишок образуются эрозии и язвы;
- увеличивается опасность желудочно-кишечных кровотечений и перфораций, что может привести к перитониту и летальному исходу.
Эндокринные нарушения возможны при воспалении сосудов (васкулите), которые вызываются непосредственно самим системным миозитом, а также в виде последствия терапии ГКС — основного метода лечения при симптомах полимиозита и дерматомиозита.
В первую очередь поражаются гипофиз, надпочечники, половые железы.
Основные методы обследования при диагностировании ДМ:
- ОАК и БХАК (общий и биохимический анализы крови;
- иммунологические анализы;
- биопсия патологических соединительных тканей (является самым верификационным методом).

Диагностическая картина при ДМ следующая:
- СОЭ и лейкоциты незначительно повышены;
- ферменты мышечных повреждений КФК (креатинфосфокиназа) и ЛДГ (лактатдегидрогеназа) могут превышать норму в десять раз;
- также повышены альдолаза, АЛТ, АСТ.
Лечение дерматомиозита
Лечить воспаление соединительных тканей при ДМ необходимо при помощи:
- глюкокортикостероидов;
- иммуносупрессантов (метотрексат, сульфасалазин, азатиоприн, циклофосфомид и др.);
- антибиотиков, антивирусных препаратов;
- антигистаминных средств.

При полимиозите, поражающем органы, проводят также комплексную поддерживающую терапию и профилактику возможных осложнений:
- Назначаются препараты для нормализации кровообращения и обмена в мягких тканях, антигипоксанты, витаминные комплексы.
- При лечении острых форм легочного полимиозита может потребоваться срочная ИВЛ, при поражении ЖКТ — экстренная операция, а при сердечной патологии — меры по предотвращению инфаркта или постинфарктная терапия.
Хирургическое вмешательство необходимо при разрастаниях соединительных тканей, их фиброзе или кальцинозе, если они затрагивают нервы, причиняя боль и ограничивая подвижность, или перекрывают сосуды.
В целом прогноз дерматомиозита благоприятен, за исключением паранеопластического и острого типа, в сочетании с поражением легких или сердца, либо приведшего к перитониту.
Дерматомиозит и полимиозит – это заболевания, характеризующиеся системным негнойным воспалением соединительной ткани, поперечно-полосатой и гладкой мускулатуры с нарушением её двигательной функции.
- При дерматомиозите (болезнь Вагнера, болезнь Вагнера-Унферрихта-Хеппа) поражение мышц сочетается с поражением кожи в виде эритемы и отека на открытых участках тела.
- При полимиозите развивается только поражение мышц, а кожный синдром отсутствует.
Заболеваемость составляет примерно 1:200000 в год. Девочки и женщины болеют в 2 раза чаще.
Этиология и патогенез
Причина заболевания неизвестна. Существует мнение, что в его развитии имеют значение вирусная инфекция и аутоиммунные реакции. В крови больных дерматомиозитом детей обнаружены антитела к пикорнавирусам, а у больных полимиозитом взрослых к Toxoplazma gondii. У больных гриппом и инфекцией, вызванной вирусом Коксаки, может развиться легкий миозит. Но до настоящего времени вирусологические и серологические исследования и отсутствие эффекта от антибактериальной терапии токсоплазмоза не подтверждают вирусную природу этих заболеваний. Но, возможно, измененные вирусы содержат антигены, напоминающие собственные антигены организма и вызывающие образование аутоантител к цитоплазматическим белкам (антигену J0-1).
Аутоиммунная природа заболеваний подтверждается обнаружением аутоантител к ядерным или цитоплазматическим антигенам, культурам эпителиальных клеток человека (Нер-2), синтетазам транспортной РНК (например, к гистидил-тРНК-синтетазе) и внутрисосудистых отложений иммунных комплексов, повреждающих эндотелиальные клетки. Иммуногистохимические исследования мышечной ткани при полимиозите выявляют воспалительные инфильтраты, содержащие активированные цитотоксические лимфоциты СD8 наряду с лимфоцитами СD4 и макрофагами. При дерматомиозите в биоптатах мышц обнаруживают отложения иммуноглобулинов и компонентов комплемента С5b–С9, а вокруг мелких сосудов накапливаются СD4 + -клетки и β-лимфоциты. Развивается поражение сосудов, опосредованное антителами и приводящее к образованию очагов некроза в мышечных волокнах, их дегенерации и потере поперечной исчерченности.
Регенерация скелетных мышц проявляется увеличением числа мышечных ядер и базофилией мышечных элементов. В последующем развивается интерстициальный фиброз.
Классификация
В настоящее время используется классификация (по R.L. Woltman, 1994), в которую вошли идиопатические воспалительные миопатии и миопатии, вызываемые инфекциями, лекарствами и токсинами.
- Идиопатические воспалительные миопатии:
- Первичный полимиозит.
- Первичный дерматомиозит.
- Ювенильный дерматомиозит.
- Миозит, ассоциирующийся с опухолями.
- Миозит с "включениями".
- Миозит, ассоциирующийся с эозинофилией.
- Локализованный или очаговый миозит.
- Оссифицирующий миозит.
- Гигантоклеточный миозит.
- Миопатии, вызываемые инфекциями.
- Миопатии, вызываемые лекарственными средствами.
Симптомы
Первичный полимиозит. Может начинаться внезапно или постепенно. Вначале развивается симметричная мышечная слабость проксимальных мышц конечностей, особенно бедер. Больным трудно подниматься и спускаться по лестнице, вставать из положения на корточках. При поражении мышц плечевого пояса больные испытывают затруднение при подъеме рук вверх и причесывании. Нередко поражаются мышцы шеи, что сопровождается нарушением ее сгибания. У многих больных развивается слабость поперечнополосатых мышц глотки и верхней трети пищевода, приводящая к дисфагии. У некоторых больных поражаются дыхательные мышцы, что приводит к тяжелым дыхательным нарушениям. Дистальные мышцы конечностей поражаются сравнительно редко. Атрофия мышц и контрактуры могут развиться на поздних стадиях заболевания. Примерно у 35% больных имеются поражения сердечной мышцы, проявляющиеся инфарктоподобными изменениями на ЭКГ, аритмиями и сердечно-сосудистой недостаточностью. У некоторых больных развиваются артралгии и синдром Рейно.
Первичный дерматомиозит. Составляет треть случаев дерматомиозита-полимиозита. Поражение мышц развивается у большинства больных одновременно с изменениями кожи. На коже появляются локальные или диффузные эритематозные участки с характерным лиловым оттенком, шелушением и атрофией. Высыпания чаще всего локализуются на веках, переносице, щеках, лбу, над локтевыми, коленными и пястнофаланговыми суставами и распространяются на шею и верхнюю часть туловища. Нередко выявляется синдром Готтрона (шелушение и атрофия кожи, наиболее выраженная на кистях и стопах), гелиотропные ("солнечные") веки (припухшие и имеющие фиолетовое окрашивание) и "руки мастерового" (эритематозные гипертрофические изменения кожи ладоней и пальцев).
Поражения суставов. Иногда у больных развиваются симметричные воспалительные поражения мелких суставов кистей и лучезапястных суставов. Но деструктивные изменения суставов обычно отсутствуют. Воспаление носит преходящий характер и быстро купируется при лечении глюкокортикоидами.
Поражения легких. Чаще всего проявляются интерстициальными пневмониями, альвеолитом и медленнопрогрессирующим фиброзом, обусловленными антителами к синтетазам транспортных РНК. Проявляются эти поражения непродуктивным кашлем и прогрессирующей дыхательной недостаточностью. Поражение диафрагмальных мышц и развивающаяся сердечная недостаточность усугубляют смешанную одышку.
Поражение почек. Развивается редко и проявляется протеинурией и нефротичексим синдромом. Иногда вследствие выраженной миоглобинурии развивается острая почечная недостаточность.
Дерматомиозит и полимиозит при злокачественных новообразованиях. Примерно у 10–25% больных выявляют онкологические заболевания, главным образом, у взрослых старше 40 лет. Особенно высок риск у онкологических больных старше 60 лет. Миопатии чаще всего развиваются при раке легкого, ЖКТ, молочной железы, матки, яичников и лимфопролиферативных заболеваниях. Данные формы дерматомиозита–полимиозита рассматривают как самостоятельные и относят к паранеопластическим синдромам. Для исключения злокачественных новообразований проводят тщательное клиническое, гинекологическое, ректальное и по показаниям рентгенологическое исследование, общий и биохимический анализ крови, цитологическое исследование мокроты и мочи. Дополнительно рекомендуют определение простатспецифического антигена у мужчин и СА-125 у женщин (антиген опухоли яичника). Прогноз неблагоприятный, больные погибают от основного заболевания, осложнений или не поддающегося лечению поражения мышц.
Ювенильный дерматомиозит. Среди больных дерматомиозитом примерно 25% составляют дети, у которых наблюдается клиническая и гистологическая картина васкулита с поражением кожи, мышц, ЖКТ, почек и других органов. Нередко развиваются очаги некроза кожи, ишемические инфаркты мышц, почек и ЖКТ. При прогрессировании заболевания развиваются контрактуры вследствие кальцификации мышц и околосуставной подкожной клетчатки. Прогноз заболевания такой же, как у взрослых. Но существует мнение, что у детей прогноз лучше, чем у взрослых. Возможно, эта противоречивость обусловлена различием диагностических критериев ювенильного дерматомиозита.
Миозит с "включениями". Данная форма относится к воспалительным миопатиям, носит спорадический характер и по клинике напоминает полимиозит. Болезнь развивается обычно у пожилых мужчин, проявляется очаговым порождением как проксимальных, так и дистальных мышц конечностей и имеет медленное прогрессирование. Характерно для миозита с "включениями" раннее появление выраженной слабости четырехглавой мышцы, сгибателей пальцев и запястья. Типичный гистологический признак, выявляемый в биоптате мышц, – вакуоли, окруженные ободком из базофильных гранул, содержащих амилоид. С помощью электронной микроскопии обнаруживают в саркоплазме и ядрах включения, напоминающие по форме нуклеокапсиды парамиксовирусов. В сыворотке крови отсутствуют аутоантитела. Характерна резистентность к глюкокортикоидам и другим иммуносупрессивным препаратам. Через 5–10 лет при хроническом прогрессировании течения больные утрачивают способность к самостоятельному передвижению.
Дерматомиозит и полимиозит, ассоциированный с ревматическими болезнями. Поражение проксимальных мышц, идентичное полимиозиту, может быть при системной склеродермии, ревматоидном артрите, системной красной волчанке и реже при узелковом периартериите и ревматизме. Но чаще всего миозит является компонентом смешанного заболевания соединительной ткани. На долю этой формы дерматомиозита–полимиозита приходится 20% всех перекрестных синдромов, для которых характерны полиартрит и феномен Рейно, высокий титр антинуклеарных аутоантител и повышение активности КФК. Биопсия мышц не всегда позволяет привести дифференциальную диагностику, так как выявляемые патологические изменения в мышцах сходны при полимиозите и других заболеваниях соединительной ткани. Как правило, хорошо поддается лечению глюкокортикоидами.
Диагностика
Для всех форм полимиозита характерно повышение активности мышечных ферментов (КФК, альдолазы, ЛДГ, АСЛ и АЛТ), выраженность которой коррелирует со степенью активности заболевания. Показатели общего анализа крови нормальные. У большинства (80–90%) больных выявляют аутоантитела к ядерным и цитоплазматическим антигенам. Наличие аутоантител к НEр-2 клеткам является стандартным диагностическим тестом.
Все воспалительные миопатии сопровождаются образованиями специфических аутоантител. К ним относятся аутоантитела к синтетазам транспортной РНК (антитела J0-1 к гистидил-тРНК-синтетазе) и распознающим сигналы частицам, взаимодействующим с нуклеопротеинами миозина. При сочетании полимиозита с системной склеродермией обнаруживают антитела к Sel-70, a с СКВ и синдромом Шёгрена – к Sm, Ro/SS-A La/SS-В.
При регистрации электромиограмм у большинства больных (80%) выявляют типичную миопатическую триаду: мелкие, короткие и полифазные (более чем из четырех фаз) потенциалы действия двигательных единиц. При гистологическом исследовании биоптата мышц выявляют очаговый некроз, дегенерацию и фагоцитоз мышечных волокон и воспалительные инфильтраты, состоящие из лимфоцитов, макрофагов и плазматических клеток. В скелетных мышцах развивается регенерация с базофилией мышечных элементов, увеличением числа и величины мышечных ядер. По периферии мышечных пучков развивается атрофия мышечных волокон, обусловленная некрозом эндотелия и уменьшением количества капилляров. При хроническом течении заболевания формируются интерстициальные фиброзные инфильтраты.
Диагностические критерии были разработаны A. Bohan et J.B. Petter в 1975 г. Эти критерии следует дополнить пунктами о наличии недеструктивного артрита и обнаружении аутоантител J0-1. При обнаружении одного типа поражения кожи и четырех других признаков из пунктов 1–4 и аутоантител J0-1 диагноз будет достоверен (чувствительность – 98,9%, специфичность – 95,2%).
Диагностические критерии дерматомиозита–полимиозита (по А. Bohan, J.B. Petter, 1975):
- Симметрическая слабость мышц плечевого и тазового пояса и передних сгибателей шеи, прогрессирующая в течение нескольких недель или месяцев в сочетании или при отсутствии дисфагии или поражения дыхательной мускулатуры.
- При гистологическом исследовании мышц признаки некроза мышечных фибрилл 1-го и 2-го типов, фагоцитоз, регенерация с базофилией, крупные ядра и ядрышки в сарколемме, перифасциальная атрофия, вариабельность размера миофибрилл, воспалительный экссудат.
- Повышение сывороточной концентрации мышечных ферментов (КФК, альдолаза, АСТ, АЛТ и ЛДГ).
- Электромиографические изменения: короткие, мелкие полифазные моторные единицы, фибрилляции и т.д.
- Дерматологические проявления, включающие гелиотропную окраску кожи век с периорбитальным отеком; чешуйчатый эритематозный дерматит на тыльной поверхности кистей, особенно над пястно-фаланговыми и проксимальными межфаланговыми суставами (симптом Готтрона) и поражение кожи над коленными, локтевыми суставами, лица, шеи, верхней половины грудной клетки.
Генерализованная мышечная слабость или слабость проксимальных мышц в сочетании с типичными кожными изменениями являются характерными признаками дерматомиозита–полимиозита. Но мышечная слабость, сопровождающаяся или не сопровождающаяся миалгиями, может быть при многих других заболеваниях.
Лечение
Основными препаратами при лечении тяжелого дерматомиозита–полимиозита являются глюкокортикоиды (преднизолон, метилпреднизолон). Начальная доза глюкокортикоидов колеблется от 1 до 2 мг/кг/сут, которую в течение 2–3 нед принимают 3 раза в день, затем – один раз в утренние часы. Улучшение наступает в течение 3–4 нед лечения, но у некоторых больных оно наступает через 3 мес. Отсутствие эффекта к этому времени являются основанием для увеличения дозы глюкокортикоидов.
После улучшения состояния больных, нормализации мышечной силы и КФК суточную дозу преднизолона уменьшают на 5 мг каждые 4 нед с обязательным регулярным контролем мышечной силы и активности КФК в сыворотке. Но следует учитывать то, что преднизолон в начале лечения может снижать активность КФК без уменьшения воспаления и инфильтрации мышц. Для уменьшения побочных эффектов больших доз глюкокортикоидов необходимо сочетать их с приемом Н 2 -блокаторов, витамина Д и препаратов кальция. После снижения суточной дозы до 40 мг переходят на прием 80 мг препарата через день. Если развивается рецидив заболевания дозу препарата вновь увеличивают до стойкого улучшения.
При неэффективности лечения в течение 3 мес высокими дозами глюкокортикоидов рекомендуют назначение азатиоприна в дозе 2–3,5 мг/кг/сут в 2–3 приема. Эффект развивается через 4–5 мес, показателем эффективности является снижение числа лимфоцитов до 750 мкл -1 . В последние годы используют метотрексат в начальной дозе 10 мг/нед с последующим ее постепенным повышением до 0,5 мг/кг/нед. Сочетанная терапия этих препаратов с глюкокортикоидами позволяет снизить дозу последних. Циклофосфамид и циклоспорин менее эффективны и их применяют при развитии интерстициального фиброза легких. Но все эти препараты угнетают кроветворение, вызывают нарушения ЖКТ и повышают риск инфекций.
По данным последних исследований, при неэффективности глюкокортикоидов улучшение может быть достигнуто с помощью в/в введения нормального иммуноглобулина в дозе 0,4 г/кг/сут в течение 5 сут. Применение плазмафереза показано больным с тяжелыми формами дерматомиозита–полимиозита, которым проводится лечение сочетанием глюкокортикоидов с азатиоприном или метотрексатом.
Очень важное значение имеет реабилитация больных с помощью физиотерапии, ЛФК и вспомогательных приспособлений. В острую фазу заболевания больной с помощью методиста выполняет пассивные упражнения и напряжение мышц, при улучшении состояния показаны изометрические и изотонические упражнения для восстановления мышечной силы.
Прогноз
Лечение большинства больных эффективно, но общая смертность при дерматомиозите-полимиозите в 4 раза выше, чем среди населения в целом. Пятилетняя выживаемость составляет 80%, у детей она еще выше.
Прогноз хуже у женщин и пожилых больных, при остром и тяжелом течении заболевания (высокая лихорадка, выраженная дисфагия, поражение внутренних органов), поздней диагностике и неадекватном лечении, наличии в сыворотке антител к J0-1 и злокачественных новообразованиях.
Читайте также:
