
Редкое генетическое заболевание костей
Генетические заболевания обусловлены патологическими нарушениями строения генома. "Дефектный" ген может быть получен от одного из родителей и проявиться как на 100%, так и на 10%. А вот болезни с наследственной предрасположенностью значительно отличаются от генетических. Если последние излечить невозможно, то заболевания, к которым человек имеет наследственную предрасположенность, возможно нивелировать рациональным питанием, здоровым образом жизни и профилактическими мерами.
Пять генетический заболеваний позвоночника и костей
Такие болезни напрямую связанны с нарушениями генома и проявляются в виде дефектов развития скелета человека. Генетические заболевания обусловлены нерациональным формообразованием ткани или нарушениями роста. Подобные болезни носят в медицине общие название - дисплазии.
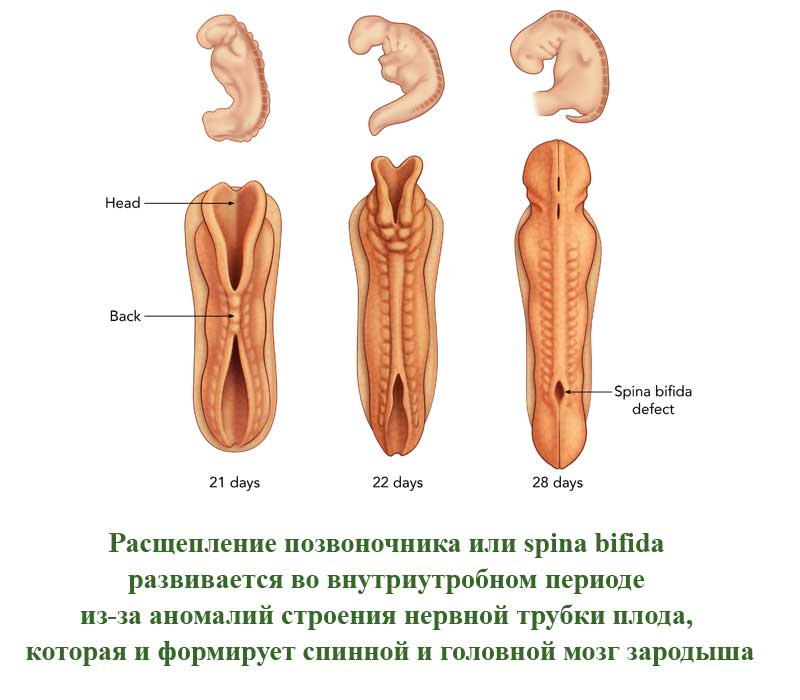
Это порок развития позвоночного столба, которое проявляется в виде недоразвитых позвонков. Такие позвонки не сомкнуты, через щель может быть виден спинной мозг. Заболевание развивается во внутриутробном периоде из-за аномалий строения нервной трубки плода, которая и формирует спинной и головной мозг зародыша. Расщепление позвоночного столба может проявляться и в закрытом виде, когда спинной мозг не виден снаружи.
В легких случаях заболевание могут обнаружить лишь при рентгеновском обследовании. А вот при самых серьезных формах болезни у ребенка могут сразу же образовываться свищи в полости позвоночника. Очень часто заболевание в тяжелых формах сопровождается параличом нижней части тела.
В более, чем 80% случаев, расщепление позвоночника сопровождается гидроцефалией спинного мозга и пороками развития головного мозга, а также - черепа.
По американской статистике, заболевание встречается у одного пациента из 1500. Российская статистика приводит следующие данные - 3 случая на 10000 человек. Однако, многие случаи расщепления позвоночника на территории СНГ остаются нераспознанными у новорожденных из-за легкой формы болезни.
Болезнь часто именуют остеопетрозом. Может протекать в двух формах:
- замедленной;
- злокачественной.
Генетическое заболевание встречается с частотой в 1 случай на 20000 пациентов. Для остеопетроза характерны такие симптомы:
- повышенная ломкость костей;
- увеличение плотности костной ткани;
- уменьшение размеров костномозговых лакун;
- нарушение гемопоэза;
- уменьшение массы костного мозга.
Генерализированный остеоклероз проявляется в достаточно раннем возрасте в виде разных беспорядочных слоев клеток костной ткани, увеличения общей массы костей и замедленном росте скелета.
При злокачественном течении болезни часто возникают внезапные переломы костей, развивается геморрагичекий синдром, жировая дистрофия органов, нарушается дентиногеез. Характерен очень небольшой рост.
В случае замедленного остеопетреза болезнь может быть выявлена лишь в 50% и протекать абсолютно бессимптомно. Выявляют заболевание случайно во время рентгена. В некоторых случаях может наблюдаться симптоматика синдрома "Кость внутри кости".
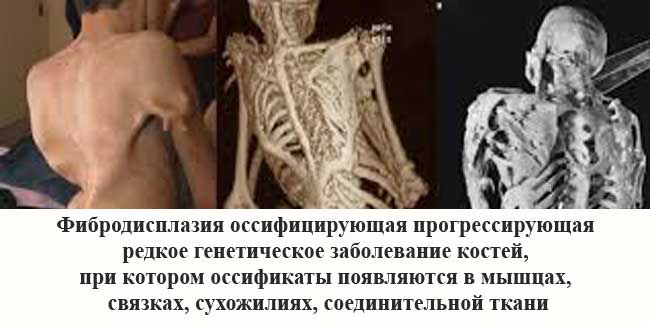
ФОП - это генетическое и очень редкое заболевание костей. При такой болезни организм начинает формировать новую костную массу в виде оссификатов в ненадлежащих местах тела, а именно внутри:
- соединительных тканей;
- связок;
- мышц;
- сухожилий.
К образованию оссификатов в организме может привести абсолютно любая травма: порез, операция, ушиб, внутримышечная инъекция или перелом. Поэтому образования такого типа удалять нельзя - на их месте костная ткань разрастется еще больше. По физиологическим признакам оссификаты совершенно не отличаются от здоровых костей.
Проблема лишь в неправильном расположении образования костной ткани. Возникает ФОП из-за мутаций гена ACVR1/ALK2. Данный ген кодирует рецептов костного морфогенетического белка. Носителем гена быть невозможно, его наличие в теле всегда вызывает развитие фибродисплазии оссифицирующей. Передается заболевание по наследству и на данный момент является неизлечимым.
Такие заболевания характеризуются чрезмерным развитием костной массы. Носят общее название - остеохондродисплазии. Гиперостозы возникают из-за генетических нарушений и патологий остеобластов и остеокластов. Наиболее часто встречаются такие формы остеохондродисплазий:
- Болезнь Лери или мелореостоз;
- пикнодизостоз.
Мелореостоз чаще всего поражает мужчин, может развиться в любом возрасте. Характеризуется болезнь избыточным образованием эндостальной или периостальной кости. Процесс может происходит в двух зонах одновременно. Зарождается болезнь Лери с поражения нижних конечностей. Процесс может переходить на все суставы, отдельные кости таза, позвоночный столб, ребра и даже череп. Все пораженные кости довольно слабо изменены и деформированы, кортикальный стой утолщен, а костномозговая полость сужена неравномерно.
Мелореостоз может протекать совершенно бессимптомно продолжительное время, однако, при значительном уменьшении габаритов костномозговых лакун развивается болевой синдром в пораженной конечности. Нога при этом может укорачиваться или увеличиваться, развивается анкилоз сустав, нарушается гемопоэз.
Пикнодизостоз проявляется в виде карликовости и остеоскрероза. В основе заболевания лежит неравномерное, чрезмерное и очаговое периостальное развитие компактной кости. Развивается явная деформация скелета в виде:
- сколиоза;
- кифоза;
- гипоплазии ключиц;
- укорочении пальцевых фаланг;
- уменьшении длины предплечий.
В молочных зубах ребенка быстро развивается кариес, склеры глаз приобретают характерных болезни голубой оттенок. На продолжительности жизни пикнодизостоз не сказывается.

Фатальная семейная бессонница
Фатальная семейная бессонница — редчайшая наследственная болезнь, при которой человек погибает от неспособности заснуть. До сих пор она отмечалась лишь в сорока семьях по всему миру. Фатальная бессонница обычно проявляется между 30 и 60 годами (чаще всего — после 50 лет) и продолжается от 7 до 36 месяцев. По мере того как заболевание прогрессирует, пациент страдает от все более тяжелых нарушений сна, причем никакие снотворные ему не помогают. На первой стадии бессонница сопровождается паническими атаками и фобиями, на второй к ним прибавляются галлюцинации и повышенное потоотделение. На третьей стадии болезни человек полностью теряет способность спать и начинает выглядеть намного старше своих лет. Затем развивается деменция, и пациент погибает — как правило, от истощения или пневмонии.
Для болезни характерен аутосомно-доминантный тип наследования: то есть у нее нет носителей. Детям она передается от родителей с вероятностью 50% и только при условии, если кто-то из них болен. Мужчины и женщины болеют фатальной семейной бессонницей с одинаковой частотой. На сегодняшний день это заболевание считается неизлечимым.

Нарколепсия-катаплексия
Примерно в 80% случаев нарколепсии сопутствует катаплексия: внезапная эпизодическая потеря мышечного тонуса, которая повторяется регулярно. В легких случаях у пациента слегка отвисает нижняя челюсть и возникает ощущении слабости в коленях, но если состояние тяжелое, человек может внезапно упасть на ровном месте. Его сознание при этом остается ясным. Катаплексия развивается на фоне выраженных эмоциональных реакций: смеха, злости, страха или удивления, что делает подобное состояние особенно неудобным.
Причина возникновения болезни пока ясна не до конца, но в ряде случаев отмечалась мутация мутация нейромедиатора орексина (ген HCRT, 17q21), который регулирует процесс передачи возбуждающих сигналов в мозгу и влияет на сон и аппетит. Сигнальная система между орексинергическими и другими нейронами дает сбой, угнетается активность моноаминергических нейронов и снижается приток возбуждающих сигналов в кору.
Устранить нарколепсию-катаплексию невозможно, однако симптоматическое лечение есть. Пациенты начинают чувствовать себя лучше благодаря регулярному сну в строго определенное время и препаратам, активизирующим работу центральных адренергических систем.

Фибродисплазия
Фибродисплазия оссифицирующая прогрессирующая (ФОП) — редкое генетическое заболевание, при котором организм начинает формировать новые кости — оссификаты — в неположенных местах: внутри мышц, связок, сухожилий и других соединительных тканей. К их образованию может привести любая травма: ушиб, порез, перелом, внутримышечная инъекция или операция. Из-за этого удалять оссификаты нельзя: после хирургического вмешательства кость может только сильнее разрастись. Физиологически оссификаты не отличаются от обыкновенных костей и могут выдерживать значительные нагрузки, вот только находятся не там, где надо.
ФОП возникает из-за мутации в гене ACVR1/ALK2, кодирующем рецептор костного морфогенетического белка. Она передается человеку по наследству от одного из родителей, если он тоже болен. Быть носителем этого заболевания нельзя: пациент либо болен, либо нет. Пока ФОП относится к числу неизлечимых болезней, однако сейчас проводится вторая серия испытаний препарата под названием паловаротен, который позволяет заблокировать ген, ответственный за патологию.

Прогерия
Детская прогерия, или синдром Хатчинсона-Гилфорда, — заболевание, которым страдает один человек из миллионов. Организм детей с таким диагнозом стареет чрезвычайно быстро и рано: уже в подростковом возрасте пациенты выглядят и чувствуют себя, как старики. У них развивается множество старческих патологий, возникают нарушения в работе внутренних органов и систем, а кости, кожа, мышцы и сухожилия становятся слабыми и вялыми. При этом по уровню развития дети с прогерией не уступают сверстникам, а иногда и опережают их. Средняя продолжительность жизни людей, страдающих синдромом Хатчинсона-Гилфорда, — 13 лет. Как правило, причиной смерти становится инфаркт миокарда. Описан всего один случай, когда пациент с таким диагнозом дожил до 45 лет.
На данный момент это заболевание также считается неизлечимым, однако продлить жизнь пациентов и улучшить ее качество помогает разнообразное симптоматическое лечение и физическая активность, особенно плавание. Эти средства позволяют улучшить состояние кровеносной системы и суставов. Кроме того, используется гормон роста.

Синдром РОХХАД
Синдромом РОХХАД (Rapid-onset Obesity with Hypothalamic dysfunction, Hypoventilation and Autonomic Dysregulation) — чрезвычайно редкое заболевание, при котором человек начинает стремительно набирать вес и страдает от булимии, респираторных болезней, остановок дыхания во сне, альвеолярной гиповентиляции и кардиореспираторных остановок. Также для пациентов с таким диагнозом характерно отсутствие реакции на повышение в крови углекислого газа.
На сегодняшний день в мире зарегистрировано порядка 100 случаев возникновения этого расстройства. Обычно оно проявляется в возрасте до 10 лет (чаще всего около 3 лет) и, по всей видимости, имеет наследственную природу. Несмотря на проведенные на Западе исследования, этиология синдрома РОХХАД до сих пор не ясна. Считается, что он появляется из-за дисфункции гипофиза, которую вызывает генетическая мутация. Однако ученым еще только предстоит определить, какой именно процесс нарушается в этом случае.
Иконки: 1) Jamie Dickinson, 2) Jaclyne Ooi, 3) Abigail Cramer, 4) Luis Martins, 5) James Stone — from the Noun Project.
Болезнь Брутона
Болезнь Брутона (первичная гипо- или агаммаглобулинемия взрослых) — наследственное заболевание семейного характера, сцепленное с Х-хромосомой, однако нередко также рецессивное наследование. Часто уже в раннем детстве отмечается склонность к повторным бактериальным инфекциям различной локализации (легкие, придаточные пазухи носа, кожа). К типичным признакам относятся диарея, а также увеличение периферических лимфатических узлов и селезенки. Возможны системные ревматические проявления по типу диффузных болезней соединительной ткани. Суставной синдром характеризуется эпизодической мигрирующей полиартралгией либо острым, подострым, но чаще хроническим моно- или асимметричным олигоартритом крупных суставов. В синовиальной жидкости признаки воспаления слабо выражены или отсутствуют вовсе. Даже при длительном течении артрит не приводит к рентгенологическим изменениям пораженных суставов. В анализах крови СОЭ в норме, острофазовые показатели не изменены, однако снижены уровень гамма-глобулинов (или они отсутствуют), иммуноглобулинов (тотально или избирательно), отсутствуют изогемагглютинины. В костном мозге выявляется отсутствие или снижение содержания плазмоцитов, а в биоптате лимфатического узла — сужение кортикального слоя, первичные фолликулы в нем редкие и малоразвитые.
Болезнь Волкова
Болезнь Гоше
Болезнь Гоше — относительно редкое заболевание, распространенное преимущественно среди евреев-ашкенази и обусловленное дефицитом лизосомальной бета-глюкоцереброзидазы. В результате происходит массивное накопление глюкоцереброзида в селезенке, печени, легких, костном мозге и других органах и тканях с образованием характерных гигантских клеток, содержащих вакуоли глюкоцереброзида (клетки Гоше). У больных, как правило, увеличены печень и селезенка, а иногда и периферические лимфатические узлы. Кроме того, выявляется очаговая кожная пигментация охряного или коричневого цвета, а у некоторых больных — желтые пятна на склерах. У детей наблюдается поражение нервной системы. В анализах крови отмечаются анемия, лейкопения и тромбоцитопения, которая может приводить к геморрагическому синдрому с носовыми кровотечениями и подкожными кровоизлияниями. В ряде случаев обнаруживается моноклоновая гаммапатия. Нередко развиваются костные повреждения (чаще всего деструкция шейки, головки или верхней трети бедра), приводящие к болям, деформации костей, патологическим переломам. Рентгенологическое исследование обнаруживает увеличение объема кости, чередование в ней участков уплотнения и разрежения либо диффузную декальцификацию костей с явлениями спонгиоза, истончение кортикального слоя, кистозные и остеосклеротические изменения. Могут возникать боли и припухлость крупных суставов, чаще нижних конечностей. Иногда поражается также поясничный отдел позвоночника, что приводит к сплющиванию (компрессии) тел позвонков. Для детской формы болезни Гоше типична быстрая смерть, при хроническом течении — медленное прогрессиро-вание. Диагноз основывается на выявлении в стернальном пунктате гигантских клеток Гоше.
Болезнь Мухи—Хаберманна
Болезнь Мухи—Хаберманна (Mucha—Habermann) — довольно распространенное, но редко распознаваемое кожное заболевание, встречающееся у детей. Характерно появление оспоподобных везикул на коже туловища, бедер, предплечий. Эти элементы могут эволюционировать в геморрагии, некроти-зироваться и разрешаются обычно с образованием корочек. Как правило, кожные поражения протекают изолированно, но иногда сопровождаются субфебрильной лихорадкой, а в ряде случаев — артритом (в том числе по типу ревматоидного артрита), склеродермией, интерстициальным пневмонитом, повышенной чувствительностью к укусу пчел. Предполагают возможную связь заболевания с реактивацией инфекции, вызванной вирусом Эпштейна—Барр.
Болезнь Тиманна
Болезнь Тиманна (Thiemann) — наследственное заболевание (аутосомно-доминантный тип наследования), встречающееся у юношей в начальном периоде полового созревания и проявляющееся множественной остеохондропа-тией (асептическим остеонекрозом) фаланг пальцев кистей, а иногда и стоп. Поражаются эпифизы средних фаланг указательного, среднего и безымянного пальцев (кроме большого пальца), обычно вовлекаются одновременно 2— 3 пальца обеих кистей, но не симметрично. Выявляется веретенообразная припухлость в области проксимальных, а иногда и дистальных межфаланго-вых суставов. Могут поражаться также межфаланговый сустав I пальца стоп и I тарзо-метатарзальный сустав. При рентгенологическом исследовании отмечается уменьшение, а нередко резорбция пораженных эпифизов, соответствующая фаланга укорачивается. Возможно спонтанное выздоровление, однако чаще остаются явления выраженного вторичного остеоартроза.
Болезнь Фабри
Болезнь Фабри (Fabry) — врожденное заболевание, характеризующееся наследственным дефицитом фермента альфа-Г4-галактозидазы, приводящим к накоплению гликолипидов (церамида) в цитоплазме и лизосомах клеток различных органов и тканей. Заболевание начинается с детства. Изменения кожи проявляются ее утолщением и ангиоэктазиями, поражение глаз — помутнением роговицы. При поражении синовиальной оболочки суставов происходит утолщение капсулы суставов и сухожильных влагалищ, отмечаются жгучие боли в пальцах кистей и стоп, голенях, предплечьях при повышении внешней температуры и ослабление болей при охлаждении конечностей. Периодически наблюдается отечность голеностопных и коленных суставов на фоне немотивированного субфебрилитета. Таким образом, клиническая картина часто напоминает воспалительное ревматическое заболевание. Рентгенологическое исследование выявляет в костях кистей множественные энтезопатические кальцификаты, а также мелкие интра- и экстраартикулярные эрозии.
Болезнь Файербанка
Болезнь Файербанка (множественная эпифизарная дисплазия) — наследственное заболевание, характеризующееся нарушением развития эпифизов, главным образом длинных трубчатых костей. Заболевание проявляется в детском и юношеском возрасте. У больных отмечаются низкий рост, нарушение походки, контрактуры и деформации преимущественно суставов нижних конечностей, широкие кисти с утолщенными и укороченными пальцами, вывихи и подвывихи в коленном и локтевом суставах, деформация стоп за счет укорочения пальцев. При рентгенологическом исследовании выявляются уменьшение размеров поперечника эпифизов, их деформация, уплощение, нередко — дольчатое строение (фрагментация). Указанные изменения носят симметричный характер и наиболее выражены главным образом в тазобедренных, коленных, плечевых и локтевых суставах.
Мукополисахаридозы
Мукополисахаридозы — группа наследственных заболеваний (аутосомно-рецессивный тип наследования), в основе которых врожденный дефект энзимов, принимающих участие в обмене глюкозаминогликанов — основных компонентов органического матрикса соединительной ткани. Отмечаются аномальная продукция, чрезмерное накопление и выделение одного или нескольких определенных мукополисахаридов, вследствие чего развиваются патологические изменения в хряще (множественный дизостоз), фасциях, периосте, сухожилиях, сердечных клапанах (сморщиваются в результате рубцовых процессов), кровеносных сосудах, черепно-мозговых оболочках, роговице, печени, селезенке (см. синдромы Марото-Лами, Моркио, Пфаундлера-Гурлера, Шийана).
Синдром Джеккейя
Синдром Джеккейя (Giaccai) — редкое семейное заболевание с аутосомно-рецессивным типом наследования, наблюдающееся в районах с высокой частотой близкородственных браков и характеризующееся прогрессирующим симметричным лизисом дистальных частей конечностей (акроостеолизом) с их последующей деформацией. Болезнь начинается в юношеском, реже — в детском возрасте и клинически похожа на остеомиелит и саркому. У больных наблюдается симметричный лизис костей дистальных отделов конечностей и последующая их деформация. На подошвенной поверхности стоп над участком рассасывания костей образуются очаги безболезненной припухлости, чаще без кожной гипертермии. Через некоторое время на этих участках появляются длительно не заживающие кожные дефекты (язвы), через которые происходит спонтанное отторжение мелких костных секвестров. После этого язвенные дефекты заживают, но могут наблюдаться и торпидно текущие.
Течение заболевания рецидивирующее с ремиссиями от нескольких недель до 3—4 лет. В итоге происходит выраженная деформация стоп с ампутацией отдельных ногтевых фаланг, деформацией и укорочением пальцев. Кроме того, отмечаются трофические изменения ногтей, гипотрофия мышц голеней и омозолелость подошвенных поверхностей, гипостезия кистей и стоп с сохранением тактильной чувствительности. Рентгенологически выявляются остеолиз фаланг и дистальных отделов плюсневых костей, очаги их деструкции без четкой периостальной реакции.
Синдром Клиппела—Тренонея
Синдром Клиппела—Тренонея — врожденная сосудистая аномалия в виде обширных варикозных расширений в области конечностей (в том числе кистей и пальцев) и верхнего плечевого пояса. Постепенно развивается гипертрофия мягких тканей конечностей, отмечается тенденция к изъязвлению кожных покровов. Происходит деформация костей (искривление, утолщение) и суставов. Возможно образование анкилозов. В области пораженных суставов могут прощупываться и рентгенологически определяться флеболиты.
Синдром Кускоквим
Синдром Кускоквим (по названию реки в штате Аляска, США, в районе дельты которой впервые были выявлены больные с данным синдромом) — комплекс наследственных аномалий (аутосомно-рецессивный тип наследования), включающий множественные контрактуры крупных суставов (преимущественно коленных и локтевых), косолапость и косорукость.
Синдром Лери
Синдром Лери — наследственное заболевание костей, характеризующееся поражением обычно одной из верхних или нижних конечностей и проявляющееся болями, распространяющимися вниз по конечности, а позже — ограничением подвижности в суставах на пораженной стороне. Часто на соответствующей стороне наблюдаются также склеродермия, атрофия и кальциноз мягких тканей. Рентгенологически обнаруживается эндостальный и частично пе-риостальный остеосклероз, возможно выявление продольных полос кальциноза в мягких тканях пораженной конечности.
Синдром Лутца—Жансельма
Синдром Лутца—Жансельма (Lutz—Jeanselme) — гиперэозинофилия в сочетании с образованием околосуставных узелков. Постепенно появляются различной величины и консистенции безболезненные подкожные узелки, локализующиеся преимущественно над костными выступами на поверхности суставов, чаще на локтях, над мелкими суставами пальцев, над областью тазобедренных суставов. В области мелких суставов кожа над узелками нормального вида, а над крупными суставами — натянутая, синюшная. Характерны артрал-гия, перемежающаяся лихорадка, слабость, кахексия. Возможно развитие неэрозивного полиартрита с вовлечением периартикулярных тканей. В крови и синовиальном выпоте определяется высокая эозинофилия. В анамнезе у больных различные аллергические заболевания. Болезнь тянется годами, обострения чередуются с частыми периодами ремиссии.
Список редких генетических заболеваний и расстройств от А до Я
Генетические заболевания присутствуют на протяжении всей жизни человека, некоторые из которых появляются очень рано. Они приводят ко многим хроническим заболеваниям, которые не поддаются лечению. Вот редкие генетические заболевания и расстройства, которые наблюдаются у людей.
Генетические заболевания или расстройства возникают из-за аномалий в генетическом составе человека. Такие аномалии могут быть вызваны незначительными, значительными изменениями или мутациями в одном или нескольких генах, хромосомными аберрациями и редко из-за мутаций в нехромосомной ДНК митохондрий. Генетические заболевания или расстройства могут быть или не быть наследственными. Они могут быть рецессивными или доминирующими по природе.
Редкая болезнь в одной части мира не может быть редкой в другой. В Соединенных Штатах критерии редких заболеваний определены в Законе о редких заболеваниях 2002 года. Он определяет такие заболевания строго в соответствии с его распространенностью, а именно «любое заболевание или состояние, которым страдают менее 200 000 человек в Соединенных Штатах, или около 1 в 1500 году. "Существует около 6000 известных генетических заболеваний, многие из которых являются дегенеративными, изнурительными и часто смертельными. В этой статье давайте узнаем о некоторых более редких формах генетических заболеваний и расстройств.
Синдром Айкарди
Синдром Айкарди - это очень редкое генетическое заболевание, характеризующееся недоразвитостью или отсутствием мозолистого тела, структуры, разделяющей левую и правую половину мозга. Оказалось, что он влияет только на девочек, так как считается, что он вызван дефектом Х-хромосомы. Это затрагивает 1 из 100 000 - 150 000 человек в Соединенных Штатах. Младенцы, рожденные с этим расстройством, кажутся нормальными до возраста от 3 до 5 месяцев, и затем начинают проявляться некоторые ключевые симптомы этого расстройства.
Симптомы включают аномалии сетчатки и инфантильные спазмы, приводящие к судорогам. Люди, затронутые этой болезнью, имеют серьезные проблемы с развитием.
Лечение не существует, нет лекарства от этого расстройства. Лечение обычно включает управление судорогами и оказание поддержки пострадавшим людям за счет задержки в развитии.
Синдром Алагилла
Синдром Алагилла - это редкое генетическое заболевание, которое поражает печень, почки, сердце и другие органы тела. Симптомы, связанные с этим синдромом, обычно отмечаются в первые годы жизни. Это затрагивает приблизительно 1 из 70 000 новорожденных. Он наследуется как аутосомно-доминантный признак, и тяжесть симптомов может варьироваться от человека к человеку. Нарушение работы печени вызвано аномалиями в желчном протоке (например, меньше или отсутствует, узкий или неправильно сформированный), что приводит к накоплению желчи в печени и, таким образом, к ее повреждению.
Симптомы включают желтоватый оттенок на коже и белках глаз, накопление холестерина под кожей, зуд и т. Д. Люди, страдающие этим расстройством, также сталкиваются со многими проблемами с сердцем. У них различимые черты лица - выступающий лоб, острый подбородок и глубоко посаженные глаза.
Лечение от этого синдрома нет, как и лекарства. Однако коррекционная хирургия, направленная на функционирование сердца, печени и почек, в некоторой степени помогает.
Алкаптонурия
Алкаптонурия, также известная как болезнь черной мочи, вызвана нарушением метаболизма тирозина в организме. Это затрагивает приблизительно 1 из 250 000 - 1 миллион человек во всем мире. Это аутосомно-рецессивное состояние, характеризующееся накоплением в крови алкаптона или гомогентизиновой кислоты (токсичного побочного продукта тирозина), которая выделяется с мочой. Моча таких пациентов становится черной при воздействии воздуха. Присутствие избытка алкаптона может привести к остеоартриту, болезням сердца, камням в почках и камням простаты у мужчин.
Симптомы: Некоторые из симптомов алкаптонурии включают потемневшую кожу и пигментированную склеру (белая часть глаза).
Лечение: Особого лечения не было установлено. Однако было отмечено, что помогает употребление продуктов, богатых витамином С. Также желательно избегать диет, богатых тирозином и фенилаланином.
Синдром альстрёма
Синдром Альстрёма - это редкое аутосомно-рецессивное заболевание, вызывающее полиорганную дисфункцию. Это одно из самых редких известных генетических заболеваний, в истории которого известно всего около 500 случаев.
Симптомы: Показания включают детское ожирение, нейросенсорную тугоухость и нарушение зрения. Это также может привести к гиперинсулинемии, гипертриглицеридемии, раннему развитию диабета 2 и глухоте. Этот синдром также может вызывать некоторые опасные для жизни медицинские осложнения, связанные с поражением печени, сердца, легких и т. Д.
Лечение: от этого синдрома нет лекарства; Тем не менее, лекарства могут быть предоставлены для лечения конкретных симптомов синдрома.
Аперт синдром
Аперт синдром является редким генетическим заболеванием, которое проявляется при рождении. Это приводит к искажению формы черепа и лица, а иногда руки и ноги перепончатые. Это влияет на 1 из 65 000 до 88 000 новорожденных. У людей, пораженных этим синдромом, кости черепа преждевременно срастаются, что называется краниосиностозом, в то время как мозг продолжает развиваться внутри аномального черепа, вызывая давление на череп и лицо, что приводит к его искажению.
Симптомы: это также приводит к плохому интеллектуальному развитию человека, потере слуха, частым инфекциям уха и пазухи и низкому росту. В большинстве случаев синдром возникает в результате случайной генной мутации, а в некоторых редких случаях он наследуется как аутосомно-доминантный признак.
Лечение - это не излечивается, но хирургическое вмешательство может до некоторой степени вылечить некоторые из его симптомов.
Болезнь Баттена
Болезнь Баттена - это редкое аутосомно-рецессивное заболевание, которое в основном поражает нервную систему и начинается в детском возрасте. По оценкам, это происходит в каждых 2-4 из каждых 100 000 человек. Болезнь Баттена характеризуется накоплением пигментов, называемых липофусцинами, в клетках организма.
Симптомы. Симптомы обычно появляются в возрасте от 5 до 10 лет, когда у нормального ребенка внезапно появляются проблемы со зрением и судороги. С течением времени симптомы ухудшаются, что приводит к потере двигательных способностей, умственной отсталости, потере зрения и т. Д.
Лечение. От этого состояния не существует лекарства, и к 20 годам оно обычно приводит к летальному исходу.
Синдром Бардета-Бидля
Синдром Бардета-Бидля - это редкое генетическое заболевание, поражающее несколько органов. Поражает от 1 из 140 000 до 1 из 160 000 новорожденных. Признаки и симптомы могут отличаться среди людей, которые страдают от синдрома, даже среди членов семьи.
Симптомы: для него характерны нарушения зрения, ожирение, аномалии почек, проблемы развития, лишние пальцы рук и ног, нарушение двигательных навыков и т. Д. Это расстройство носит рецессивный характер.
Лечение: лечения от этой болезни нет, и обычно лечение концентрируется на конкретных симптомах.
Болезнь Камурати-Энгельмана
Болезнь Камурати-Энгельмана является разновидностью дисплазии кости. Это редкое аутосомно-доминантное генетическое заболевание. На сегодняшний день во всем мире зарегистрировано только 200 случаев заболевания.
Симптомы: для него характерны утолщенные кости, что приводит к хронической боли. Кости черепа также утолщаются, что приводит к давлению на мозг, что приводит к различным неврологическим проблемам. Люди, страдающие от этого, также жалуются на повышенную утомляемость, слабость, головную боль и мышечные спазмы.
Лечение: от этого состояния нет лекарства, но его можно частично лечить. Противовоспалительные и иммунодепрессивные агенты, такие как глюкокортикостероиды, оказались полезными в некоторых случаях. Также рекомендуются альтернативные методы лечения, такие как массаж и тепловая терапия в сочетании с медикаментозным лечением.
Синдром Карпентера
Синдром Карпентера - это редкое врожденное заболевание, характеризующееся неправильной формой головы, лица, пальцев рук и ног из-за преждевременного сращения костей. Это аутосомно-рецессивное заболевание с примерно 100 зарегистрированными случаями, известными до настоящего времени. По оценкам, это влияет на 1 из 1000 000 человек.
Симптомы: Основные симптомы включают головку странной формы, слитые цифры, ожирение, низкий рост и снижение умственных способностей. 50% детей с этим расстройством имеют порок сердца, который осложняет ситуацию.
Лечение: Лечение обычно состоит из ряда поэтапных операций, направленных на исправление пороков развития костей на ранних этапах жизни. Это состояние похоже на синдром Аперта и синдром Пфейффера.
Семейные идиопатические базальные ганглии кальцификации
Ранее известный как синдром Фара, это редкое генетически доминирующее заболевание. Характеризуется аномальными отложениями кальция в базальных ганглиях и коре головного мозга. Это области управления движением мозга.
Симптомы: Ключевыми симптомами являются неуклюжесть, усталость, неустойчивая осанка, мышечные спазмы, неконтролируемые движения и деменция. Люди могут быть затронуты этим синдромом в любое время их жизни; однако, это более распространено в возрастной группе 40-50 лет. Известно, что только 60 семей страдают от этого синдрома в медицинской литературе.
Лечение: от этого состояния нет лекарства. Лечение обычно симптоматическое.
Наследственный ангионевротический отек
Наследственный ангионевротический отек, также известный как болезнь Квинке, вызван нарушениями функции белка, называемого ингибитором С1. Мутации в гене ингибитора C1 (C1NH) приводят к HAE. Это влияет на кровеносные сосуды, что приводит к проблемам иммунной системы. По оценкам, это происходит в 1 из 50000 человек.
Симптомы: Общие симптомы наследственного ангионевротического отека включают отек рук, ног, глаз и горла, боли в животе и закупорку дыхательных путей.
Лечение: Лечение обычно проводится после приема лекарств, гормонального лечения и введения обезболивающих препаратов.
Болезнь Хантингтона
Болезнь Хантингтона является аутосомно-доминантным генетическим заболеванием и более распространена в зрелом возрасте. Со временем симптомы усугубляются, и пострадавшему человеку требуется постоянный уход. Это условие влияет на 1 из 10000 в Соединенных Штатах. Ожидаемая продолжительность жизни после начала заболевания составляет 15-25 лет.
Симптомы: Показатели включают непроизвольные движения, потерю двигательных способностей, когнитивные трудности и эмоциональные проблемы.
Лечение: лечения не существует, но лекарства могут облегчить определенные симптомы, связанные с расстройством.
Синдром Меккеля
Синдром Меккеля - чрезвычайно редкое фатальное генетическое заболевание. Он наследуется по аутосомно-рецессивному типу и поражает примерно 1 из 13 250-140 000 человек во всем мире. Из-за его серьезных симптомов люди обычно умирают во время родов или вскоре после рождения. Это чаще встречается в Бельгии и Финляндии, где примерно 1 из 3000-9000 человек страдает от этого.
Симптомы : у него тяжелые признаки и симптомы, включая уродливую центральную нервную систему, многочисленные заполненные жидкостью кисты в почках, дефекты легких, печени, полидактилию.
Лечение : может быть рекомендовано восстановление сердца или нейрохирургическое вмешательство при энцефалоцеле.
Мукополисахаридоз VI
Мукополисахаридоз VI - это генетическое заболевание, которое встречается у 1 из 300 000 новорожденных. Это вызвано дефицитом активности N-ацетилгалактозамин-4-сульфатазы (арилсульфатазы В), которая ответственна за катаболизм сложных углеводов (полисахаридов). Это приводит к накоплению дерматансульфата в некоторых органах, таких как скелет, легкие, клапаны сердца, селезенка и печень.
Симптомы: Симптомы включают низкий рост, умственную отсталость, респираторные заболевания, глухоту и слепоту.
Лечение: Лечение проводится после заместительной энзимотерапии и регулярной медицинской помощи.
Синдром Пфейффера
Синдром Пфайффера связан с мутацией рецептора фактора роста фибробластов, который важен для нормального развития костей. Это редкое аутосомно-доминантное генетическое заболевание, поражающее 1 человека на 100 000 человек. Характеризуется аномальным слиянием костей черепа, что приводит к деформации головы и лица. Это также может повлиять на кости ног и рук.
Симптомы: Видимые симптомы этого синдрома: необычно широкий лоб, выпученные глаза, клюв носа, короткие широкие пальцы рук и ног. В некоторых случаях также видна перепонка цифр. Индивидуум, страдающий от этого синдрома, также сталкивается с потерей слуха и зубными проблемами.
Лечение: поэтапные черепно-лицевые операции обычно проводятся в первые месяцы жизни, чтобы исправить деформацию костей.
Прогерия
Прогерия - это редкое генетическое заболевание, которое встречается у 1 из 8 000 000 живорожденных. Это генетическое заболевание, вызванное новой мутацией в гене и, как правило, не наследуемое. Это вызывает быстрое старение у детей, и в результате люди, затронутые этой болезнью, умирают в возрасте от 13 до 20 лет.
Симптомы: Ключевыми симптомами прогерии являются проблемы роста на первом году жизни, морщинистое маленькое лицо, большая голова, потеря волос, бровей и ресниц, плохое зрение и снижение моторных навыков. Генетический тест на мутацию LMNA может подтвердить, есть ли у человека прогерия.
Лечение: от этой болезни нет лекарства. Все предлагаемые варианты лечения, как правило, носят поддерживающий характер и зависят от симптомов.
Синдром Ретта
Синдром Ретта - это редкое генетическое нарушение развития нервной системы. Это аутосомно-доминантное Х-сцепленное расстройство, которое чаще встречается у женщин, чем у мужчин. Это затрагивает 1 из 10000 человек. Симптомы обычно появляются после 6-18 месяцев жизни.
Симптомы: они включают проблемы с дыханием, сенсорные проблемы, неустойчивую походку, неспособность к обучению, судороги, социальную неловкость или безответственность. Симптомы часто неверно интерпретируются как аутизм и церебральный паралич.
Лечение: от этого синдрома нет лекарства; однако исследования в этой области продолжаются.
Дефицит рибозо-5-фосфат-изомеразы
Дефицит возникает из-за мутации в энзиме рибозо-5-фосфат-изомеразы, которая играет жизненно важную роль в пентозофосфатном пути. Только с одним диагностированным случаем это расстройство считается самым редким. МРТ показала высокий уровень арабита и рибита, что указывает на врожденную ошибку метаболизма полиолов. У пациента были такие симптомы, как болезнь белого вещества и невропатия неизвестного происхождения. Причину этого состояния еще предстоит полностью понять. Есть несколько теорий, делающих раунды, но этот недостаток все еще изучается.
Болезнь Сандхоффа
Болезнь Сандхоффа - это редкое генетическое нарушение накопления липидов, которое разрушает нервные клетки головного и спинного мозга. Это вызвано из-за дефицита функциональных бета-гексозаминидаз A & B, которые приводят к отложению определенных липидов в мозге и других органах тела. Это аутосомное расстройство затрагивает 1 из 300 000 человек. Из 3-х типов этого синдрома классический инфантильный является самым летальным.
Симптомы: У пораженных детей проявляются мышечная слабость, потеря двигательных навыков, страдают потерей слуха, параличом, потерей зрения, пятнами красной вишни, умственными нарушениями и обычно выживают до возраста 3-4 лет.
Лечение: Лечение этого состояния, как правило, является поддерживающим.
Туберозный склероз
Туберозный склероз ранее был известен как болезнь Борнвилля. Это редкое генетическое заболевание, которое поражает примерно 1 из 10 000 человек во всем мире. Около 80% случаев связаны с мутациями в двух специфических генетических локусах - TSC1 и TSC2. Туберозный склероз вызван ростом опухолевидных опухолей головного мозга, легких, сердца и почек.
Симптомы: Общие симптомы включают поражения кожи в области носа и щек, периунгулярную фиброму, эпилептические припадки, поведенческие проблемы, заболевания легких и почек и умственную отсталость.
Лечение: Симптомы эпилептических припадков можно лечить хирургическим путем, удаляя клубни, которые отвечают за эпилепсию. Проводятся исследования, чтобы найти лекарственную терапию для этого заболевания.
Болезнь Вольмана
Болезнь Вольмана - это редкое генетическое заболевание, поражающее примерно 1 из 35 000 человек. Это аутосомно-рецессивное заболевание, вызванное дефицитом лизосомально-кислой липазы (LAL). LAL - это фермент, необходимый для расщепления определенных липидов в клетках. У людей, страдающих от этого состояния, вредное количество липидов накапливается в печени, селезенке, лимфатических узлах, костном мозге и других частях тела. Симптомы обычно проявляются в первые несколько недель жизни.
Симптомы: к ним относятся проблемы с кормлением, постоянная рвота, диарея, увеличение печени и селезенки, плохое увеличение веса и т. д.
Лечение : от этого состояния не существует лекарства; Однако существуют методы лечения, которые являются симптоматическими и поддерживающими.
Синдром Циммерманна-Лабанда (ZLS)
Только с 44-мя известными случаями синдром Циммермана-Лабанда нам известен. Он является очень редким врожденным аутосомно-доминантным заболеванием.
Симптомы: для него характерен фиброматоз десен (крупные десны), отсутствие или недоразвитие ногтей конечных фаланг пальцев рук и ног, аномальный нос и ухо и грубый внешний вид лица.
Лечение: Лечение этого синдрома обычно симптоматическое и поддерживающее.
Генетические заболевания и расстройства являются серьезной проблемой для медицинских исследователей во всем мире. Исследования по клонированию генов, генной терапии и подавлению генов, ответственных за возникновение генетических заболеваний и замену ферментов, все еще находятся в поисках способов лечения редких генетических заболеваний и расстройств.
Читайте также:
