
Растяжки для детей с мышечной дистрофией дюшена болеют мальчики
Мышечная дистрофия Дюшенна, МДД - страшный лотерейный билет. Его может вытащить любая пара , которая планирует стать родителями, независимо от расы, образа жизни и социального статуса.
- Расскажите, что такое мышечная дистрофия Дюшенна?
- Мышечная дистрофия Дюшенна (МДД)– редкое генетическое заболевание, которым в 99% случаях болеют мальчики. У больных с диагнозом МДД не вырабатывается белок дистрофин в гене, который отвечает за формирование мышечной ткани. С ростом ребенка происходит постепенное разрушение всех мышц в организме, в частности и сердца, которое тоже является мышцей. Отказывают все двигательные системы – трудно ходить, кашлять, дышать и так далее. Заболевание редкое и считается самым финансово затратным в мире. Случаев один на четыре тысячи. На Западе люди с диагнозом МДД могут доживать до 50 лет и не выживать, как у нас, а именно жить! Получать образование, заводить семьи и самое главное иметь здоровых детей. В России диагноз МДД – приговор!
- Приговор?!
- Да! У нас ребята с МДД доживают до двадцати лет, в лучшем случае…
- Почему?
- Нет нужных препаратов, и даже их разработки не ведутся. Западные же лекарства нельзя ввозить в Россию, та как их нет в реестре разрешенных препаратов. Да и стоить они будут от 600 000 евро в год. От куда у нас такие деньги?! А такие препараты отдельной категории детей смогли бы уже существенно замедлить прогрессирование заболевания. Нашим детям нужен дистрофин, как например диабетикам инсулин.
- Действительно нет российских аналогов?
- Неужели до сих пор никто не обратил внимания на эту проблему?
- Можете привести пример?
- Например, ребенок уже сам давно не ходит и ему требуется откашливатель и аппарат для искусственной вентиляции легких, а ему предоставляют ортопедическую обувь….
- На какие симптомы стоит обратить внимание родителям мальчиков?
- Примерно до пятилетнего возраста ребенка, врачи и родители находятся в полной уверенности, что малыш здоров. Ребеночек растет и вдруг начинает спотыкаться падать на ровном месте, дальше изменяется походка, может отмечаться хождение на носках. Ему трудно вставать с пола – поднимается с помощью опоры. Обратить внимание следует на увеличение икроножных мышц. Если они стали неестественно крупные, то это возможно признак начала мышечного распада. Дальше появляется трудности при хождении, ребенок не может подниматься по лестнице встать с пола без помощи, откашливании, меняется осанка, что значительно ухудшает качество жизни.
Меньше чем за десять лет здоровый на вид ребенок, садится в инвалидное кресло (болезнь быстро прогрессирует). Примерно к 17 годам мы имеем беспомощного молодого человека, который требует постоянного и дорогостоящего ухода…
- А дальше?
- Что происходит с родителями, когда они узнают о диагнозе ребенка?
Родители впадают в глубокую депрессию, порой рушится семья, уходят папы, родители вынуждены уйти с работы, чтобы осуществлять уход за ребенком, большинству требуется психологическая помощь…
Тяжело это. Очень!
- Я знаю, что у Вас есть сын…
- Да, его зовут Никита.
- Как Вы справляетесь, с его диагнозом?
- Все слезы уже выплаканы… Я поняла, что рассчитывать не на кого. Я должна жить, чтобы мой сын мог жить. В какой-то степени придает сил наша Общественная Организация. Мы - Родительский проект и всегда морально поддерживаем друг друга. Объединились больше 350 семей со всей России, в которых ребенок страдает МДД, чтобы начать вести диалог с врачами, обществом и Государством о проблемах, которые у нас есть.

Сын Елены Шевкуновой Никита. Мальчик хорошо учится в школе а на досуге любит слушать джаз Фото из семейного архива Елены Шевкуновой
- Какие ваши главные цели и задачи?
- Чтобы дети не умирали в 9-14 лет! Мы понимаем, что наши дети уже не увидят разработок российских препаратов от МДД. Пока эти лекарства откроют, пока их испытают, одобрять их еще дольше будут - пройдет десятилетие, как минимум. Но нам нужна хотя бы нормальная поддерживающая и своевременная медицина! На уход в год нужно примерно 800 000 рублей… И чтобы врачи были грамотней в этом вопросе, чтобы общество знало о проблеме. Такой ребенок может родиться в каждой семье! Обязательно нужно проводить семинары на эту тему, делать ДНК - диагностики мамам и скрининг детям.
Наше сообщество родителей даже как-то назвали интеллектуальными волонтерами, которые занимаются миссионерской деятельностью!
- Существует какой-то способ узнать, что человек может попасть в группу риска?
- Да. Носителем является женщина, у носительниц может быть повышен уровень КФК (креатинкиназа) – это фермент, который необходим каждой клетке в организме. Особенно важен этот фермент в клетках нервной ткани и мышц. У детей с МДД этот показатель, завышен в десятки раз.
ВАЖНО
+7 958 630 81 91
КСТАТИ
Почему родители таких малышей рискуют оказаться под следствием (подробности)

Мышечная дистрофия Дюшенна – это генетическое заболевание, связанное с нарушением строения мышечных волокон. Мышечные волокна при этой болезни, в конце концов, распадаются, и теряется способность к передвижению. Мышечная дистрофия Дюшенна передается сцеплено с полом, болеют лица мужского пола. Проявляет себя уже в детском возрасте. Помимо мышечных нарушений, заболевание приводит к скелетным деформациям, может сопровождаться дыхательной и сердечной недостаточностью, умственными и эндокринными нарушениями. Радикального способа лечения, позволяющего искоренить болезнь, пока нет. Все существующие меры являются лишь симптоматическими. Довольно редко больным удается пережить рубеж 30 лет. Эта статья посвящена причинам, симптомам, диагностике и лечению мышечной дистрофии Дюшенна.
Заболевание впервые описано в 1861 году (по другим данным – 1868 году) французским невропатологом и носит его имя. Встречается не так уж и редко: 1 случай на 3500 новорожденных детей. Из всех известных медицине мышечных дистрофий является наиболее распространенной.
Причина заболевания

В основе мышечной дистрофии Дюшенна лежит генетический дефект половой Х хромосомы.
Один из участков Х хромосомы содержит ген, кодирующий производство в организме особого мышечного белка под названием дистрофин. Белок дистрофин составляет основу мышечных волокон (миофибрилл) на микроскопическом уровне. Функция дистрофина заключается в поддержании клеточного скелета, в обеспечении способности миофибрилл к многократным актам сокращения и расслабления. При мышечной дистрофии Дюшенна этот белок либо отсутствует вообще, либо синтезируется дефектным. Уровень нормального дистрофина не превышает 3%. Это приводит к разрушению мышечных волокон. Мышцы перерождаются и заменяются жировой и соединительной тканью. Естественно, что при этом утрачивается двигательный компонент человеческой деятельности.
Симптомы заболевания
Мышечная дистрофия Дюшенна всегда заявляет о себе до 5-летнего возраста. Наиболее часто первые симптомы возникают еще до 3-х лет. Все патологические проявления заболевания можно разделить на несколько групп (в зависимости от характера изменений):
- поражение скелетной мускулатуры;
- деформации скелета;
- поражение сердечной мышцы;
- умственные нарушения;
- эндокринные расстройства.

Поражение мышечной ткани является основным проявлением заболевания. Оно становится причиной генерализованной мышечной слабости. Начальные симптомы подкрадываются незаметно.
Рождаются дети без особых отклонений. Однако их двигательное развитие отстает в темпах по сравнению со сверстниками. Такие дети менее активные и подвижные в двигательном плане. Пока ребенок совсем мал, это часто связывают с особенностями темперамента и не обращают внимания на начальные изменения.
Явные признаки возникают с началом ходьбы. Дети часто падают и передвигаются на пальцах (на носочках). Следует отметить, что эти нарушения интерпретируют не при первых шагах ребенка, потому что прямохождение вначале для всех детей сопряжено с падениями и неуклюжестью. Когда большинство ровесников уже вполне уверенно передвигается, мальчики с мышечной дистрофией Дюшенна упорно продолжают падать.
Когда ребенок научится разговаривать, он начинает жаловаться на слабость и быструю утомляемость, непереносимость физических нагрузок. Бег, лазанье, прыжки и другие любимые виды активной деятельности детей для ребенка с мышечной дистрофией Дюшенна не привлекательны.
Походка таких детей напоминает утиную: они как бы переваливаются с ноги на ногу.
Мышечная дистрофия Дюшенна имеет восходящий тип мышечной слабости. Это означает, что сначала слабость проявляет себя в ногах, затем распространяется на таз и туловище, потом на плечи, шею и, в конце концов, на руки, дыхательную мускулатуру и голову.
Несмотря на то, что при этом заболевании мышечные волокна подвергаются разрушению и развитию атрофии, внешне некоторые мышцы могут выглядеть вполне нормальными или даже накаченными. Развивается так называемая псевдогипертрофия мышц. Чаще всего этот процесс заметен в икроножных, ягодичных и дельтовидных мышцах, мышцах языка. Место распавшихся мышечных волокон занимает жировая ткань, именно поэтому создается эффект хорошей развитости мускулатуры, что на проверку оказывается совсем не так.
Разрушение мышц сопровождается развитием мышечных контрактур и укорочением сухожилий (хорошо заметно на примере ахиллового сухожилия).
Сухожильные рефлексы (коленный, ахиллов, с бицепса, с трицепса и так далее) постепенно снижаются. Мышцы на ощупь плотные, но безболезненные. Мышечный тонус обычно снижается.
Постепенное прогрессирование мышечной слабости приводит к тому, что к 10-12 годам многие дети утрачивают способность самостоятельно передвигаться и нуждаются в инвалидном кресле. Способность стоять сохраняется, в среднем, до 16 лет.
Отдельно следует сказать о вовлечении в патологический процесс дыхательной мускулатуры. Это наблюдается после подросткового периода. Слабость диафрагмы и других мышц, участвующих в акте дыхания, приводит к постепенному снижению жизненной емкости легких и объемов вентиляции. По ночам это особенно заметно (появляются приступы удушья), поэтому у детей могут возникать страхи перед сном. Формируется дыхательная недостаточность, которая усугубляет течение интеркуррентных инфекций.

Это сопутствующие мышечным изменениям симптомы. У детей постепенно формируются усиление поясничного изгиба (лордоза), искривление грудного отдела позвоночника в сторону (сколиоз) и сутулость (кифоз), меняется форма стопы. Со временем развивается диффузный остеопороз. Эти симптомы еще больше способствуют ухудшению двигательных нарушений.
Является обязательным симптомом мышечной дистрофии Дюшенна. У больных развивается кардиомиопатия (гипертрофическая или дилатационная). Клинически это проявляет себя нарушениями сердечного ритма, перепадами артериального давления. Границы сердца увеличиваются, однако такое большое сердце имеет малые функциональные возможности. В конце концов, формируется сердечная недостаточность. Сочетание выраженной сердечной недостаточности с дыхательными расстройствами на фоне присоединившейся инфекции может быть причиной смертельного исхода у больных с мышечной дистрофией Дюшенна.
Это не обязательный, но возможный признак заболевания. Он связан с дефицитом особой формы дистрофина – аподистрофином, содержащимся в головном мозге. Нарушения интеллекта варьируются от незначительных до степени идиотии. При этом выраженность умственных нарушений никоим образом не связана со степенью мышечных расстройств. Социальная дезадаптация из-за невозможности свободно передвигаться и посещать детские учреждения (садики, школы) способствует усугублению когнитивных расстройств.
Встречаются у 30-50% больных. Могут быть довольно разнообразными, но чаще всего это ожирение с преимущественным отложением жира в области молочных желез, бедер, ягодиц, плечевого пояса, недоразвитие (или нарушение функции) половых органов. Больные часто имеют низкий рост.
Мышечная дистрофия Дюшенна неуклонно прогрессирует. К 15-20 годам почти все больные не в состоянии себя обслуживать самостоятельно ввиду обездвиженности. В конце концов, присоединяются бактериальные инфекции (органов дыхания и мочевыделительной сферы, инфицированные пролежни при недостаточном уходе), которые на фоне сердечной и дыхательной недостаточности приводят к смертельному исходу. Мало кто из больных переживает рубеж в 30 лет.
Диагностика

Диагностика мышечной дистрофии Дюшенна основывается на нескольких видах исследований, основным из которых является генетический тест (ДНК-диагностика).
Только обнаружение дефекта Х хромосомы в том участке, который отвечает за синтез дистрофина, достоверно подтверждает диагноз. До проведения такого анализа диагноз является предварительным.
Из других методов исследования могут применяться:
- определение активности креатинфосфокиназы (КФК). Этот фермент отражает гибель мышечных волокон. Его концентрация при мышечной дистрофии Дюшенна превышает норму в десятки и сотни раз до 5-летнего возраста. Позже уровень фермента постепенно снижается, потому что часть мышечных волокон уже необратимо разрушена;
- электромиография. Этот метод позволяет подтвердить тот факт, что в основе заболевания лежат первичные изменения мышц, а нервные проводники при этом совершенно интактны;
- биопсия мышц. С ее помощью определяют содержание белка дистрофина в мышце. Однако в связи с усовершенствованием генетической диагностики в последние десятилетия эта травматичная процедура отошла на второй план;
- дыхательные пробы (исследование жизненной емкости легких), ЭКГ, УЗИ сердца. Эти методы не используются для установления диагноза, но необходимы для выявления патологических изменений со стороны дыхательной и сердечно-сосудистой систем для того, чтобы скорригировать имеющиеся нарушения.
Выявление больного ребенка в семье означает, что в генотипе матери имеется патологическая Х хромосома. В редких случаях мать может быть здоровой, если мутация возникла у ребенка случайно. Наличие дефектной Х хромосомы несет в себе риск для последующих беременностей. Поэтому такие семьи должен консультировать генетик. При наступлении повторных беременностей родителям предлагают пренатальную диагностику, то есть исследование генотипа еще не родившегося ребенка с целью исключения наследственных заболеваний, в том числе мышечной дистрофии Дюшенна.
Для исследования понадобятся клетки плода, которые получают с помощью различных процедур на разных сроках беременности (например, биопсия хориона, амниоцентез и другие). И хотя эти медицинские манипуляции несут в себе определенный риск для беременности, они позволяют точно ответить на вопрос: имеется ли у плода генетическое заболевание.
Лечение
Мышечная дистрофия Дюшенна является в настоящее время неизлечимым заболеванием. Можно помочь ребенку (взрослому) продлить время двигательной активности, с помощью различных способов поддерживая мышечную силу, компенсируя изменения со стороны сердечно-сосудистой и дыхательной систем.
Несмотря на это, прогнозы ученых в отношении полного излечения от этого заболевания довольно оптимистичны, поскольку уже сделаны первые шаги в этом направлении.
В настоящее время, для лечения мышечной дистрофии Дюшенна из медикаментозных препаратов используют:
- стероиды (при регулярном применении они позволяют уменьшить мышечную слабость);
- β-2-адреномиметики (также временно придают силу мышцам, но не замедляют прогрессирование заболевания).
Применение β-2-адреномиметиков (Альбутерол, Формотерол) не имеет статистически достоверного признания, поскольку существует небольшой опыт их использования при данной патологии. Контроль изменений состояния здоровья у группы пациентов, применявших эти препараты, проводился в течение одного года. Поэтому утверждать, что они работают более длительное время, нет возможности.
Основой лечения сегодня являются стероиды. Считается, что их использование позволяет какое-то время сохранять мышечную силу, то есть они могут затормозить прогрессирование болезни. Кроме того, доказано, что стероиды уменьшают риск возникновения сколиоза при мышечной дистрофии Дюшенна. Но все же возможности этих препаратов ограничены, и заболевание будет неуклонно прогрессировать.
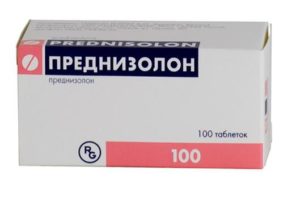
Когда же начинают лечение гормонами? Считается, что оптимальным временем для начала терапии является такая фаза заболевания, когда двигательные навыки не улучшаются, но еще не ухудшаются. Обычно это бывает в возрасте 4-6 лет. Наиболее часто используются такие препараты, как Преднизолон и Дефлазакорт. Дозы назначаются индивидуально. Препараты используются, пока есть видимый клинический эффект. Когда же наступает фаза прогрессирования заболевания, то необходимость применения стероидов отпадает, и их постепенно (!) отменяют.
Из медикаментозных препаратов также при мышечной дистрофии Дюшенна применяют сердечные средства (антиаритмические, метаболические, ингибиторы ангиотензинпревращающего фермента). Они позволяют бороться с кардиологическими аспектами заболевания.
Ортопедические приспособления позволяют значительно облегчить жизнь больного. Их перечень довольно широк и разнообразен: это и различного рода вертикализаторы (помогают сохранять положение стоя), и приспособления для самостоятельного вставания, и инвалидные кресла-коляски с электрическим приводом, и специальные шины для устранения контрактур в голени (используются даже ночью), и корсеты для позвоночника, и длинные шины для ног (колено-голеностопные ортезы), и многое другое.
Когда заболевание поражает и дыхательные мышцы, и самостоятельное дыхание становится неэффективным, то возможно применение аппаратов искусственной вентиляции легких различной модификации.
И все же даже использование всех этих мер в комплексе не позволяет побороть заболевание. На сегодняшний день, есть ряд перспективных направлений исследований, которые, возможно, станут прорывом в лечении мышечной дистрофии Дюшенна. К наиболее распространенным среди них относят:
Каждая из новых разработок несет в себе надежду для больных с мышечной дистрофией Дюшенна на полное выздоровление.
Таким образом, мышечная дистрофия Дюшенна – это генетическая проблема лиц мужского пола. Болезнь характеризуется прогрессирующей мышечной слабостью из-за разрушения мышечных волокон. В настоящее время является неизлечимым заболеванием, однако многие ученые мира трудятся над созданием радикального способа борьбы с ним.

Мышечные дистрофии – это группа заболеваний не воспалительного характера, характеризующиеся прогрессивным течением без патологических изменений центральной и периферической нервной системы.
- Что это такое?
- Этиология, кто наследует и почему
- Симптомы мышечной дистрофии
- Формы миопатии
- Методы диагностики
- Лечение заболевания
- Последствия и осложнения
- Прогноз на выздоровление и жизнь
- Что нужно запомнить?
Что это такое?
Первые публикации о миопатиях датируются 1830 годом. В 1852 году Мерион сообщил о случае в семье с четырьмя сыновьями, у которых отмечались признаки двигательных расстройств, не связанных с поражением нервной системы. Он предположил, что заболевание наследуется от матери к ребенку.
Под миопатией или миодистрофией Дюшенна (также ее называют дистрофией или миопатией Дюшена-Беккера) понимают заболевание генетической природы, связанное с мутацией гена, кодирующего синтез белка дистрофина.
Дистрофин входит в состав мембран мышечных клеток, без него мышцы крайне уязвимы и подвергаются некрозу и дегенерации. Экспрессия, то есть реализация функции, происходит в гладкой и скелетной мышечной ткани, а также в миокарде.
Миопатия Дюшенна поражает одного из 3500 новорожденных детей. Несмотря на тот факт, что заболевание названо именем Дюшенна, одним из первых врачей, описавших данную патологию, был Говерс.
Гийом Дюшенн был французским неврологом, который использовал электростимуляцию в лечении неврологических расстройств. В 1868 году Дюшенн описал 13 пациентов с заболеванием, которое сопровождалось прогрессирующей мышечной слабостью, и назвал ее паралитической псевдогипертрофической мышечной дистрофией. Также он определил диагностические критерии миодистрофии, которые используются и сегодня.
Этиология, кто наследует и почему
По международной классификации болезней МКБ-10 мышечная дистрофия Дюшенна-Беккера имеет код G71.
Одна треть случаев заболевания связана со спонтанно возникшей мутацией, в то время как на остальные случаи патологии приходится наследование Х-хромосомы, несущей патологический ген.
Случаи гонадного мозаицизма связаны с 20% случаев миодистрофии Дюшенна. Средняя продолжительность жизни менее 20 лет. Повышение уровня фермента креатинфосфокиназы (КФК) обнаруживают у 2/3 женщин – носителей патологического гена. Большинство из них не имеют никаких проявлений заболевания. Миопатия почти всегда поражает мальчиков, поскольку заболевание сцеплено с Х-хромосомой, тип наследования – рецессивный. Локализация гена, кодирующего синтез дистрофина, находится в локусе Xq21. Синтез белка кодируется одним из самых больших известных генов. Он занимает около 2% ДНК Х-хромосомы.
Симптомы мышечной дистрофии
Первые симптомы появляются в возрасте от трех до семи лет. Обычно родители замечают раскачивающуюся походку и гиперлордоз. Существует несколько основных критериев, которые позволяют предположить миопатию Дюшенна. К ним относятся:
- Мышечная слабость, которая появляется неожиданно и начинается с нижних конечностей.
- Гиперлордоз – выраженный изгиб позвоночника кпереди, что особенно проявляется при ходьбе.
- Гипертрофия ослабленных мышц.
- Слабый ответ мышц на электрическую стимуляцию на более поздних стадиях заболевания.
С течением времени все указанные симптомы прогрессируют. Несмотря на поражение мышечных структур, нарушений функции мочевого пузыря и кишечника не отмечается. К двенадцати годам жизни большинство пациентов не могут ходить самостоятельно, и находится в инвалидном кресле. Миопатия Беккера очень сходна с дистрофией Дюшенна, так как поражается один и тот же локус Х-хромосомы, в результате чего страдает синтез дистрофина. Отличием миопатии Беккера является начало заболевания, как правило, после трех лет жизни или даже в подростковом возрасте.
Миодистрофия Дюшенна – патология, которая затрагивает не только скелетную мускулатуру. Дистрофин также содержится в миокарде, тканях мозга и гладкомышечной мускулатуре. Поздняя стадия заболевания ассоциируется с тяжелой сердечной недостаточностью и дыхательными нарушениями – главными причинами смерти пациентов.
Формы миопатии
В течении заболевания различают пять стадий. Первая стадия (доклинических проявлений) не характеризуется широким спектром симптомов, обычно у пациентов отмечается лишь повышение уровня КФК в сыворотке крови.
Вторая стадия (ранних признаков) включает следующие симптомы:
- Раскачивающаяся походка – впервые появляется от 2-х до 6-ти лет и часто является первым симптомом, который замечают родители.
- Прогрессирующая слабость мускулатуры нижних конечностей с дальнейшим присоединением слабости мышц шеи, плечевого пояса и рук.
- Наличие симптома Говерса – при попытке встать с пола, пациент упирается на колени и руки (рисунок 1). Появление симптома связано с выраженной слабостью мышц спины и конечностей.

Рисунок 2. Симптом Говерса – пациент поднимается с пола, опираясь на колени и руки.
Третья стадия (прогрессирующих симптомов) характеризуется появлением значительных сложностей во время ходьбы и развивается, когда возраст ребенка составляет около восьми лет. Пациенту становится сложно подниматься по ступеням, появляется одышка, сложно встать с пола. По утрам могут беспокоить головные боли, в ночное время наблюдается затрудненное дыхание. Четвертая и пятая стадии являются наиболее тяжелыми, так как пациент теряет возможность не только самостоятельно передвигаться, но и испытывает трудности с дыханием. При четвертой стадии пациент еще может удерживать осанку, однако делать это становится все труднее, развивается выраженный сколиоз.
Пятая стадия – терминальная. Пациент не может ходить и, как правило, находится в инвалидном кресле. Чаще всего, смерть наступает от дыхательной и сердечно-сосудистой недостаточности в возрасте до 20-ти лет.
Методы диагностики
Ребенок с миопатией Дюшенна-Беккера до 2-3-х лет жизни может ничем не отличаться от других детей. Тем не менее, существует ряд признаков, на которые стоит обратить внимание.
Важно! Во время проведения лабораторных исследований может быть повышен уровень печеночных ферментов: аланин- и аспартатаминотрансферазы, креатинкиназы и гамма-глутаматтрансферазы.
В ряде исследований миопатий было отмечено, что задержка речи и моторных навыков отмечалась чаще у детей с дефектом дистрофина. Уровень IQ может быть меньше на одно стандартное отклонение по сравнению со среднестатистическим значением в популяции.
У 30% детей с мутациями гена, кодирующего синтез дистрофина, отмечались трудности с обучением и приобретением новых навыков, обсессивно-компульсивные расстройства, синдром дефицита внимания, задержка умственного развития. Детям с дистрофиями Дюшенна и Беккера сложнее даются вербальные навыки.
- Исследование уровня КФК, которая превышает референсные значения в 50-100 раз.
- Молекулярную диагностику – исследование гена, локализованного в локусе Xq21 и ответственного за синтез дистрофина, позволяет подтвердить или опровергнуть диагноз.
Если у пациента наблюдается сочетание высоких показателей КФК и мышечная слабость, это с высокой вероятностью позволяет заподозрить миопатию Дюшенна-Беккера.
Из инструментальных методов используют:
- обзорную рентгенографию грудной клетки. Исследование позволяет сделать заключение, насколько выражен сколиоз;
- электромиографию: метод применяют с целью дифференциальной диагностики со спинальной мышечной атрофией;
- электрокардиография – с целью выявления синусовых аритмий;
- ультразвуковое исследование сердца – нередко определяет малые размеры желудочков сердца и более длинную диастолу;
- холтеровское мониторирование определяет наличие пароксизмальных аритмий.
Если молекулярная диагностика не выявила мутации гена дистрофина, рекомендуется провести биопсию мышечной ткани. Характерные гистологические изменения приведены ниже:
- мышечные волокна с выраженным дегенеративным процессом и некрозом;
- пролиферация соединительной ткани;
- появление жировой ткани в значительном количестве.
Также проводится анализ белка дистрофина, выделенного из мышцы, с определением его молекулярной массы.
Лечение заболевания
На сегодняшний день лечения миодистрофии Дюшенна-Беккера не существует. Терапевтическая тактика основана на поддерживающем медикаментозном лечении (в основном, для сохранения сердечной функции), исследованиях в области генной инженерии и экспериментальном клеточном лечении.
При прогрессировании сколиоза и контрактур суставов возможно применение паллиативного хирургического вмешательства. По мере прогрессирования мышечной слабости, следует обеспечить пациенту максимальный комфорт.
Применение преднизолона активно обсуждается в профессиональных медицинских сообществах. Известно, что спустя один месяц от начала применения наблюдается незначительное улучшение общего состояния пациента и может сохраняться до трех лет без ухудшения. В то же время, при отказе от преднизолона начинается прогресс заболевания.
Последствия и осложнения
Главными осложнениями, с которыми сталкиваются пациенты, являются:
- Прогрессирующая мышечная слабость.
- Дилатационная кардиомиопатия.
- Дыхательные нарушения, связанные с дисфункцией диафрагмы.
- Контрактуры суставов.
- Сколиоз.
- Дисфагия.
- Запоры.
К осложнениям также относится остеопороз и высокий риск переломов. По некоторым данным возможно использование препаратов кальция и витамина Д для увеличения плотности костной ткани.
Прогноз на выздоровление и жизнь
К сожалению, прогноз неблагоприятный, так как смертность при данной патологии составляет 100%. Если в семейной истории встречается близкая родственница, которая является носителем патологического гена, у детей мужского пола выше риск развития кардиомиопатии с прогрессирующей сердечной недостаточностью в возрасте от 20 до 40 лет. Проводятся исследования, связанные с применением стволовых клеток для лечения миодистрофий.
Что нужно запомнить?
- Миопатия Дюшенна-Беккера – прогрессирующее заболевание, связанное с аномальным синтезом белка дистрофина, принимающего непосредственное участие в мышечных сокращениях. Синтез дефектного белка приводит к дегенеративным и некротическим процессам в мышцах.
- Ранними признаками болезни считаются появление мышечной слабости в нижних конечностях, раскачивающейся, нетипичной походки. К лабораторным признакам относится многократное повышение уровня КФК.
- Этиология заболевания – генетическая, связанная с мутацией гена в локусе Тип наследования-рецессивный, мутация может передаваться от матери, чья Х-хромосома несет патологический ген, а может возникать спонтанно.
- Существует пять стадий миопатии. Четвертая и пятая стадии являются терминальными.
- К ранним методам диагностики относится определение уровня КФК, который может многократно превышать норму и являться ранним маркером наследственной миодистрофии Дюшенна-Беккера задолго до появления мышечной слабости и других заметных симптомов. К более точным методам относится молекулярная диагностика с исследованием патологического гена.
- Специфическое лечение не разработано. Мероприятия по уходу за пациентом сводятся к поддержанию жизнеобеспечения.
- Основными осложнениями являются утрата способности ходить, тяжелые дыхательные расстройства и сердечно-сосудистая недостаточность.
- Прогноз неблагоприятный, летальность составляет 100%.
Литература
- Bushby K, Straub V. Nonmolecular treatment for muscular dystrophies. Curr Opin Neurol. 2005 Oct. 18(5):511-8.
- Moxley RT 3rd, Ashwal S, Pandya S, Connolly A, Florence J, Mathews K, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2005 Jan 11. 64(1):13-20.
- Ervasti JM, Campbell KP. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991 Sep 20. 66(6):1121-31.
- Ozawa E, Noguchi S, Mizuno Y, et al. From dystrophinopathy to sarcoglycanopathy: evolution of a concept of muscular dystrophy. Muscle Nerve. 1998 Apr. 21(4):421-38.
- Darke J, Bushby K, Le Couteur A, McConachie H. Survey of behaviour problems in children with neuromuscular diseases. Eur J Paediatr Neurol. 2006 May. 10(3):129-34.
- Mendell JR, Shilling C, Leslie ND et al. Evidence based path to newborn screening for Duchenne Muscular Dystrophy. Ann Neurology. 2012. 71:304–313.
- Rodino-Klapac LR, Chicoine LG, Kaspar BK, Mendell JR. Gene therapy for duchenne muscular dystrophy: expectations and challenges. Arch Neurol. 2007 Sep. 64(9):1236-41.
- Merlini L, Gennari M, Malaspina E et al. Early corticosteroid treatment in 4 duchenne muscular dystrophy patients: 14-year follow-up. Muscle Nerve. 2012 Jun. 45(6):796-802.
- American Academy of Pediatrics Section on Cardiology and Cardiac Surgery. Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics. 2005 Dec. 116(6):1569-73.
- Colan SD. Evolving therapeutic strategies for dystrophinopathies: potential for conflict between cardiac and skeletal needs. Circulation. 2005 Nov 1. 112(18):2756-8.
Читайте также:
