
Нервно мышечное заболевание мышечная дистрофия что это

- Атрофия мышц
- Изменение походки
- Изменение размеров мышц
- Отсутствие боли
- Слабость в ногах
- Снижение мышечного тонуса
- Усталость
- Утрата физических навыков
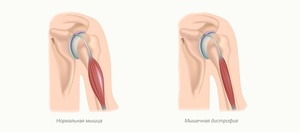
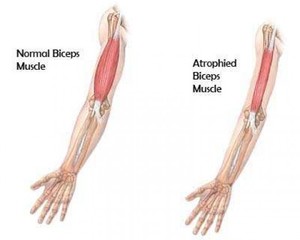
Мышечная дистрофия является группой хронических заболеваний мышечных структур, преимущественно скелетных. Для всех прогрессирующих мышечных дистрофий характерной чертой является постепенно проявляющаяся слабость мышц, а также их дегенерация. По мере развития недуга наблюдается уменьшение диаметра мышечных волокон. Поражённые элементы в результате дистрофии утрачивают свою способность сокращаться и постепенно распадаются. Их место в теле больного человека занимает соединительная и жировая ткань.
- Этиология
- Разновидности
- Симптоматика
- Диагностика
- Осложнения
- Лечение
Клиницисты выделяют всего девять разновидностей этого патологического состояния, которые имеют существенные различия в зависимости от агрессивности развития, основных характеристик, локализации поражённых волокон, а также возрастных показателей.
Этиология
Пока ещё учёным точно не удалось выяснить истинные причины, которые запускают патологические механизмы, провоцирующие патологию. Но точно уже известно, что основа всех причин этого патологического состояния – мутации аутосомно-доминантного генома, основной функцией которого является синтезирование и регенерация в теле человека специфического белка, отвечающего за полноценное формирование мышечных волокон.
В зависимости от того, какая именно хромосома в генетическом коде человека была подвержена процессу мутации, зависит, недуг с каким расположением будет развиваться:
- в большинстве клинических ситуаций мутации подвергается Х хромосома в геноме человека. В этом случае начинает прогрессировать мышечная дистрофия Дюшена. Именно эта форма недуга диагностируется чаще всего. Примечателен тот факт, что если представительница прекрасного пола имеет в своём геноме дефектную хромосому, то велика вероятность того, что она передаст её и своим потомкам. Но бывает и так, что у неё никаких симптомов недуга проявляться и вовсе не будет;
- причиной мотонической разновидности недуга является формирование аномального генома, относящегося к 19 хромосоме;
- отдельно стоит выделить мышечную недоразвитость, которая совершенно не связана с аномалиями в половой хромосоме. В данную группу относят 2 вида болезни: плечо-лопатка-лицо, поясница-конечности.
Разновидности
Клиницисты выделяют несколько наиболее распространённых форм недуга.
Мышечная дистрофия Дюшена. Данную разновидность также именуют в медицинской литературе псевдогипертрофической. Обычно мышечная дистрофия Дюшена начинает прогрессировать уже в детском возрасте. Примечателен тот факт, что этому недугу более подвержены маленькие мальчики, нежели девочки.
Прогноз этого типа дистрофии не является благоприятным – многие люди погибают, даже не достигнув 20-летнего возраста.

Болезнь Штейнерта. Эта разновидность патологии является характерной для взрослых людей из возрастной категории от 20 до 40 лет. Редки случаи, когда патология проявляет себя уже в младенческом возрасте. Ограничений, касательно половой принадлежности, дистрофия не имеет. Характеризуется медленным прогрессированием.
Дистрофия этого вида имеет свою характерную черту – патологический процесс затрагивает не только мышцы скелета, но также и структуры жизненно важных органов. У больного возможно появление слабости мышц лица. Стоит отметить, что поражение других групп мышц также не исключено. Характерно медленное расслабление волокон после их предварительного сокращения.
Прогрессирующая мышечная дистрофия Беккера. Эта разновидность патологии является малораспространенной. Патологический процесс прогрессирует довольно медленно. Прогрессирующую мышечную дистрофию Беккера обычно диагностируют у людей с низким ростом. Прогноз недуга благоприятный. На протяжении многих лет люди с таким диагнозом сохраняют свою работоспособность, и их состояние остаётся удовлетворительным. Инвалидизации способствуют травмы различной степени тяжести, а также сопутствующие болезни.
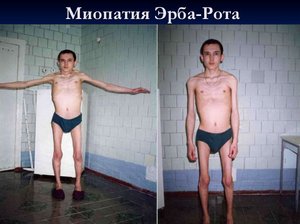
Юношеская мышечная дистрофия Эрба-Рота. Период проявления её симптомов – от 10 до 20 лет. Прогрессирует медленно. На начальных стадиях развития отмечается атрофия волокон рук и плеч, позже – ног и таза. Во время ходьбы можно отметить изменение осанки человека – грудная клетка отодвигается немного назад, в то время как живот выпячивается вперёд. Больной идёт и переваливается.

Мышечная дистрофия Ландузи-Дежерина. Симптомы недуга проявляются в период от 6 до 52 лет. Чаше всего признаки дистрофии Ландузи-Дежерина выявляют в период с 10 до 15 лет. При этом недуге в первую очередь страдают мышцы лица. Но постепенно при дистрофии Ландузи-Дежерина патологический процесс охватывает также мышечные структуры конечностей и туловища.
Первым симптомом развития болезни является неполное смыкание век. Постепенно перестают полностью смыкаться губы, что обуславливает нарушение дикции. Патология Ландузи-Дежерина у больных протекает достаточно медленно. У пациента длительное время сохраняется способность двигаться, поэтому он может вести нормальный образ жизни. В среднем через 20–25 лет возможно атрофирование мышц тазового пояса, что и становится причиной инвалидизации. В целом можно сказать, что дистрофия Ландузи-Дежерина протекает благоприятно.
Симптоматика
Различные формы дистрофии (Ландузи-Дежерина, Дюшена, Беккера и прочее) имеют свои характерные признаки прогрессирования. Но также существует группа симптомов, которые являются характерными для любой разновидности патологии:
- отсутствие болевого синдрома;
- постепенное понижение тонуса мышечных волокон;
- чувствительность в поражённых участках не понижается;
- скелетные мышцы постепенно атрофируются;
- перемена походки;
- частые падения вследствие слабости мышц ног;
- постоянная усталость;
- характерный симптом прогрессирования недуга – изменение размеров мышц;
- мышечная дистрофия у детей проявляется постепенным утрачиванием физ. навыков, которые они уже успели развить до момента прогрессирования патологии.
Диагностика
Диагностика мышечной дистрофии у детей и взрослых включает в себя такие мероприятия:
- тщательный сбор анамнеза;
- электромиография;
- взятие небольшого участка мышечного волокна для проведения микроскопического исследования;
- дополнительное консультирование у ортопеда и терапевта.
Осложнения
- нарушения работы сердца;
- деформация позвоночного столба;
- снижение интеллекта, функций памяти;
- снижение способность совершать активные движения и постепенна инвалидизация;
- прогрессирование патологий системы дыхания;
- летальный исход (медицинская статистика такова, что этот недуг редко становится причиной смерти).
Лечебные мероприятия
Стоит сразу отметить, что мышечную дистрофию излечить полностью возможности нет. Ещё не было создано такого препарата или процедуры, которые смогли бы восстановить поражённые участки мышечных волокон. Лечение мышечной дистрофии в первую очередь направлено на торможение активного развития процессов дистрофии в мышечных структурах. С этой целью больному назначают кортикостероиды, витамины, АТФ и прочее.
- лечебный массаж;
- физиотерапию;
- дыхательную гимнастику;
- проводят профилактику прогрессирования сколиоза.

Мышечная дистрофия (МД) — это группа заболеваний, характеризующихся прогрессирующей слабостью и мышечной дегенерацией. Мышцы постепенно атрофируются — теряют свой объем и, следовательно, силу.
Это болезни генетического происхождения, которые могут возникать в любом возрасте: с самого рождения, в детстве или во взрослой жизни. Существует более 30 форм заболеваний, которые различаются по возрасту появления симптомов, характеру пораженных мышц и степени тяжести. Большинство типов дистрофий постепенно осложняются и имеют необратимые последствия. В настоящее время лечения МД все еще не существует. Наиболее известным и распространенным типом заболеваний является миопатия Дюшенна.
В ходе развития МД страдают в первую очередь основные мышцы, которые способствуют произвольному движению, включая мышцы, бедра, ног, рук и предплечья. В некоторых случаях могут быть затронуты респираторные мышцы и сердце. Люди с мускульной дистрофией постепенно теряют свою мобильность при ходьбе. Другие симптомы могут быть связаны с мышечной слабостью, включая сердце, желудочно-кишечные, глазные проблемы.
Распространенность заболевания
Миопатия относится к редким и неизлечимым заболеваниям. Трудно вывести точную статистику, поскольку она объединяет различные болезни. Согласно некоторым исследованиям, около 1 из 3 500 человек страдают от этого заболевания.
Например:
![]()
Миопатия Дюшенна затрагивает приблизительно одного ребенка (мальчика) из 3500.- Миопатия Беккера касается 1 мальчика из 18 000.
- Фазио-скапулогумаральная дистрофия поражает около 1 из 20 000 взрослых людей.
- Болезнь Эмери-Дрейфус затрагивает 1 из 300 000 человек, вызывает ретракцию сухожилия и нарушение сердечной мышцы
Частота и тип заболеваний зависит от конкретной страны:
Причины заболевания и лечение
Причиной данной патологии являются генетические заболевания, то есть дефект (или мутация) гена, необходимого для нормального развития мышц. Когда этот ген мутирует, мышцы больше не в состоянии нормально функционировать — они теряют свой силовой потенциал и в результате атрофируются.
Например:
- Миопатия Дюшенна связана с дефицитом дистрофина — белка, расположенного под мембраной мышечных клеток, который играет роль в сокращении мышц.
- Почти у половины врожденных МД причиной является дефицит мерозина — белка, составляющего мембрану клеток мышцы.
Как правило, МД передается рецессивно. Другими словами, для того чтобы болезнь выражалась, оба родителя должны быть носителями и передавать ребенку ненормальный ген. Болезнь не проявляется у родителей по той причине, что у каждого из них есть только один аномальный ген, а не два. Для нормального функционирования мышц достаточно одного нормального гена.
Кроме того, некоторые формы миопатии затрагивают только мальчиков: это миопатия Дюшенна и Беккера. В обоих случаях ген, участвующий в этих двух заболеваниях, расположен в Х-хромосоме, которая существует в единственной копии у мужского пола.
Симптомы заболевания
МД проявляются мышечной слабостью, которая имеет тенденцию к постепенному ухудшению, симптомы варьируются в зависимости от типа патологии. В зависимости от случая могут присутствовать и другие симптомы, такие как сердечные и респираторные расстройства, аномалии глаз (пороки развития, катаракта), интеллектуальный дефицит, гормональные нарушения и т. д.
Дети часто жалуются на судороги и мышечные боли. Болезнь развивается довольно быстро, как только появляются первые симптомы. Обычно использование инвалидной коляски требуется примерно в возрасте 12 лет. Такого рода нарушения приводят к сколиозу и деформациям суставов. Кроме того, у некоторых детей наблюдается умственная отсталость. К концу подросткового возраста часто возникают сердечные осложнения (сердечная недостаточность), а также респираторные проблемы, требующие искусственной подачи воздуха. Средняя продолжительность жизни (от 20 до 30 лет в среднем).
Миопатия Беккера. Симптомы сравнимы с симптомами М. Д. Дюшенна , однако они менее выражены, а развитие заболевания происходит медленнее. Симптомы начинаются в 5−15 лет, иногда позже, характеризуются прогрессирующей потерей силы мышц в конечностях и в окрестностях туловища. В более чем половине случаев ходьба остается возможной до возраста 40 лет.
Миопатия Штейнтера. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Также наблюдается Миотония — аномальное и продолжительное сокращение мышц (мышца расслабляется слишком медленно), особенно выражается в руках, а иногда и на языке. Также могут быть затронуты мышцы лица, шеи и лодыжек. Часто присутствуют сердечные и дыхательные нарушения, которые являются потенциально серьезными. Нередко наблюдаются пищеварительные, гормональные, глазные расстройства, а также бесплодие и раннее облысение.
Миопатия поясничного отдела. Симптомы обычно проявляются в детстве (10 лет) или в раннем взрослом возрасте (около 20 лет). Мышцы плеч и бедер постепенно ослабевают, в то время как мышцы головы, шеи и диафрагмы обычно не затрагиваются. Если некоторые формы сопровождаются дыхательными нарушениями, то при этом типе дистрофии такие аномалии отсутствуют. Сердечные нарушения встречаются редко. Эволюция (развитие заболевания) очень изменчива, в зависимости от формы.
Миопатия Дежерина-Ландузи или плечелопаточная дистрофия. Симптомы обычно появляются в позднем детстве или в зрелом возрасте (от 10 до 40 лет). Как следует из названия, миопатия затрагивает мышцы лица, плеч и рук. Таким образом, больному становится сложно выразить улыбку, произнести некоторые предложения и закрыть глаза. Потеря подвижности происходит примерно в 20% случаев. Заболевание развивается медленно, продолжительность жизни нормальная.
Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Окуло-глоточная миотония. Это заболевание относительно распространено в Квебеке. Симптомы обычно появляются около 40 или 50 лет. Первые признаки болезни проявляются опустившимися веками, за которыми следуют слабость мышц глаз, лица и горла (глотки), вызывая трудности с глотанием пищи. Прогрессирование заболевания происходит медленно.
Исследования и прогресс
С 2005 года для лечения пациентов с развивающимся поражением мышц все чаще используются стволовые клетки. Для лечения мышечной дистрофии этим методом могут быть рассмотрены различные варианты заболевания, такие как: мышечные дистрофии Дюшенна, Беккера, миопатия поясничного и плечевого отдела.
Целью лечения является регенерация потерянных и поврежденных мышечных волокон с использованием регенеративного потенциала стволовых клеток. Для этого большое количество стволовых клеток вводится при помощи нескольких внутривенных и внутримышечных инъекций, что позволяет лучше нацеливать терапию именно на пораженную группу мышц.
Возможный прогресс
Терапия с применением стволовых клеток может обеспечить улучшение в плане мышечной массы, силы, движений, баланса, тремора и ригидности мышц. Стволовые клетки также могут замедлить будущую потерю мышечного объема и уменьшить симптомы.
Важно отметить, что лечение не является окончательным лекарством от этого заболевания и никоим образом не может решить проблему потери мышечных волокон. По этой причине прогресс после такого лечения не может быть постоянным. Исследования в этой области все еще ведутся.
Семейства заболевания
Обычно существуют два основных семейства МД:
- Мышечна врожденная дистрофия (ВМД), которая выражается в первые 6 месяцев жизни, сопровождает около десяти форм патологий переменной тяжести, включая ВМД с первичной недостаточностью мерозина, синдром Ульриха и Уокера-Варбурга;
- Мышечные дистрофии, появляющиеся в детстве или в зрелом возрасте:
![]()
Миопатия Дюшенна- Миопатия Беккера
- Миопатия Эмери-Дрейфуса (существует несколько форм)
- миопатия Ландузи-Дежерина
- Так называемая миопатия поясничного отдела — затрагивает мышцы вокруг плеч и бедер.
- Миотонические дистрофии (типы I и II), которые включают болезнь Штейнтера. Они характеризуются миотонией — когда мышцы не могут нормально расслабиться после сокращения.
- Окулофарингеальная миопатия
Эволюция дистрофии
Эволюция (развитие заболевания) МД сильно варьируется от одной формы к другой, а также от одного человека к другому. Некоторые формы быстро развиваются, что приводит к ранней утрате подвижности и ходьбе, а иногда и к смертельным сердечным или респираторным осложнениям, в то время как другие развиваются очень медленно — в течение десятилетий. Большинство врожденных мышечных дистрофий, например, которые мало выражены или почти незаметны, позже могут проявятся внезапно и с серьезными последствиями.
Возможные осложнения
Осложнения сильно различаются в зависимости от типа патологии. Некоторые нарушения могут затрагивать респираторные мышцы или сердце, иногда с очень тяжелыми последствиями.
Таким образом, сердечные осложнения довольно распространены, особенно у мальчиков с мышечной дистрофией Дюшенна.
Кроме того, дегенерация мышц заставляет тело и суставы деформироваться постепенно: на фоне этого у больных может развиваться сколиоз. Часто наблюдается сокращение мышц и сухожилий, что приводит к их стягиванию. Все эти нарушения приводят к деформации суставов: ноги и руки повернуты внутрь и вниз, деформируются колени или локти.
Также известно, что болезнь сопровождается тревожными или депрессивными расстройствами, поэтому больным требуется много внимания и поддержки, в первую очередь со стороны близких.

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Хроническая болезнь наследственного генезиса, выраженная дегенерацией мышц, поддерживающих костный каркас скелета – это мышечная дистрофия.
Медицина классифицирует девять разновидностей данной патологии, различающихся по локализации нарушения, его характеристиках, агрессивность прогрессирования, возрастным показателям пациента (сколько лет было пациенту, когда начали проявляться первые симптомы патологии).

[1], [2], [3], [4], [5], [6], [7]
Причины мышечной дистрофии
На сегодняшний день медицине не может назвать все механизмы, которые запускают процесс, приводящий к мышечной дистрофии. Однозначно можно сказать только, что все причины мышечной дистрофии базируются на мутациях аутосомно-доминантного генома, который ответственен в нашем организме за синтез и регенерацию белка, который и участвует в формировании мышечных тканей.
В зависимости от того, какая из хромосом в коде человека подверглась мутации и зависит, патологию какой локализации мы и получим на поверку:
- Мутация половой Х хромосомы приводит к наиболее распространенному виду патологии мышечная дистрофия Дюшена. Если женщина является носительницей данной хромосомы – она, зачастую передает его своим потомкам. При этом сама такими нарушениями может не страдать.
- Мотоническая мышечная дистрофия проявляется, если дефектным становится геном, принадлежащий девятнадцатой хромосоме.
- Не зависит от патологии половой хромосомы и такая локализация мышечной недоразвитости: поясница – конечности, а так же плечо – лопатка-лицо.
[8], [9], [10], [11]
Симптомы мышечной дистрофии
Симптомы мышечной дистрофии имеют комплекс основных, базовых, проявлений, но и в зависимости от локализации и характеристик патологии имеются и собственные отличительные особенности. •
- В связи с дефицитом мышечной массы ног, наблюдаются нарушения в походке человека.
- Уменьшается тонус мышц.
- Атрофируются скелетные мышцы.
- Утрачиваются двигательные способности, которые были приобретены больным до момента начала прогрессирования заболевания: пациент перестает держать голову, ходить, сидеть, теряет и другие навыки.
- Притупляются болевые ощущения мышц, при этом чувствительность не пропадает.
- Снижение общего жизненного тонуса, больной начинает очень быстро утомляться.
- Мышечные волокна начинают замещаться соединительной тканью, что приводит к увеличению объемов самих мышц. Особенно это заметно по икроножному отделу.
- Проявляются трудности к обучению.
- Достаточно часты падения.
- Возникают сложности при беге и прыжках.
- Больному становиться тяжело вставать, как из лежачего положения, так и сидячего.
- Походка такого больного становится переваливающейся.
- Идет снижение интеллекта.
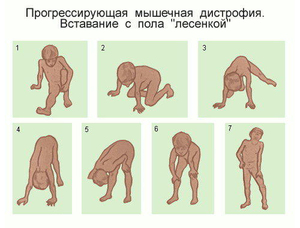
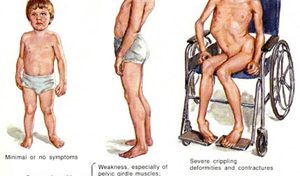
Первые признаки заболевания начинают проявляться в ослаблении тонуса нижних конечностей, а так же области таза. При дальнейшем прогрессировании заболевания подключаются к атрофии группы мышц верхней части тела. Постепенно, за счет перерождения мышечных волокон в соединительные, увеличиваются в объемах икроножные области нижних конечностей больного, увеличиваются размеры и жировой ткани. Темпы развития данного генетического нарушения достаточно велики и уже к 12 годам ребенок утрачивает способность вообще двигаться. Зачастую до двадцати лет такие пациенты не доживают.
Ослабление тонуса мышц нижних конечностей с разрастанием объемов икроножной области и приводит к тому, что ребенок начинает первично испытывать дискомфорт при ходьбе и беге, а впоследствии теряет эту способность полностью. Постепенно поднимаясь вверх и захватывая все большее количество групп мышц, на терминальной стадии мышечной дистрофии дюшена, патология начинает поражать и комплекс дыхательных мышц, глотки и лица.
Псевдогипертрофия может прогрессировать не только в икроножной области, она способна захватить и зоны ягодиц, живота и языка. При такой патологии достаточно часто возникает поражение и сердечных мышц (изменения идут по типу кардиомиопатии). Нарушается сердечный ритм, тона становятся глухими, само сердце увеличивается в размерах. Сердечная мышечная дистрофия, зачастую, и является причиной смерти пациента.
Характерной симптоматикой является и то, что больной страдает и умственной отсталостью. Это объясняется поражениями, которые захватывают и большие полушария головного мозга. При прогрессировании мышечной дистрофи, начинают появляться и другие сопутствующие заболевания. Такие, например, как: диффузный остеопороз, заболевания, связанные с эндокринной недостаточностью, идет деформация грудной клетки, позвоночника…
Основным отличительным признаком патологии по типу Дюшена от остальных видов, является большой уровень гиперферментемии, проявляющийся уже на начальной фазе развития патологии.
[12], [13], [14], [15]
Наиболее часто, в области мышечно-неврологических заболеваний, встречается первичная прогрессирующая мышечная дистрофия, которая представлена достаточно обширной классификацией. Отличие одной формы от другой идет в зависимости от места генной мутации, скорости прогрессирования, возрастной характеристикой пациента (в каком возрасте начала проявляться патология), присутствует ли в симптоматике псевдогипертрофия и другие признаки. Большая часть из этих миодистрофий (их симптоматика), за практически вековую историю, достаточно неплохо изучена, но до сих пор патогенез так и не известен, а, исходя из этого, возникают проблемы и с высокой достоверностью диагностики. Не зная причин возникновения патологических изменений, очень тяжело и провести достаточно рациональную классификацию прогрессирующей мышечной дистрофии.
В большинстве своем, деление проводится либо по форме наследования, либо по клиническим характеристикам.
Первичная форма - повреждение мышечной ткани, при которой остаются действующими периферийные нервы. Вторичная форма – когда поражение начинается из нервных окончаний, изначально не затрагивая мышечные слои материю.
- Тяжелый тип псевдогипертрофии Дюшена.
- Реже встречающийся, менее агрессивный тип Беккера.
- Тип Ландузи - Дежерина. Затрагивает область плечо-лопатка-лицо.
- Тип Эрба – Рота. Подростковая форма заболевания.
Это основные типы мышечной дистрофии, которые диагностируются наиболее часто. Остальные разновидности встречаются реже и являются атипичными. Например, такие как:
- Дистрофия Ландузи Дежерина.
- Дистрофия Эмери Дрейфуса.
- Конечностно - поясная мышечная дистрофия.
- Окулофарингеальная мышечная дистрофия.
- А так же, некоторые другие.
Эта патология встречается относительно редко и, в отличие от тяжелой злокачественной формы Дюшена, является доброкачественной и прогрессирует достаточно медленно. Одним из характерных признаков может служить то, что данной формой, как правило, болеют люди, имеющие небольшой рост. Достаточно длительное время болезнь не дает о себе знать и человек живет обычной жизнью. Толчком к развитию заболевания может явиться либо банальная бытовая травма, либо сопутствующее заболевание.
Мышечная дистрофия беккера относится к более легким формам данного заболевания и по остроте клинической симптоматики, и по полноте молекулярных проявлений. Симптомы в случае диагностики мышечной дистрофии по форме Беккера выявляются слабо. Больной с такой патологией способен достаточно нормально жить не один десяток лет. При такой слабой симптоматике дистрофию по Беккеру низко квалифицированный врач вполне может спутать с конечностно-поясничной дистрофией. Первые признаки данной патологии обычно начинают проявляться в двенадцатилетнем возрасте. Подросток начинает ощущать боли в нижних конечностях (в области голени), особенно во время нагрузки. Анализ мочи показывает высокое содержание миоглобина, являющегося показателем того, что в организме проходит распад мышечного белка. Идет повышение креатинкиназа в организме больного (фермента, вырабатывающегося из АТФ и креатина). Он активно используется организмом при возрастании физических нагрузок на него.
Эта форма мышечной дистрофии была описана врачами и учеными Беккером и Кинером еще в 1955 году, поэтому и носит их имя (ее знают как мышечную дистрофию Беккера или Беккера-Кинера).
Симптоматика выявления патологии, как и в случае болезни по форме Дюшена, начинается с отклонений в тазово-поясной области, захватывая и нижние конечности. Это проявляется в изменении походки, появляются проблемы с подъемом по лестнице, очень тяжело такому больному становиться вставать из сидячего, на низких поверхностях, положения. Постепенно увеличиваются размеры икроножных мышц. При этом изменения области ахилловых сухожилий, заметные при патологии Дюшена, в данном случае визуализируются незначительно. Не наблюдается и снижения интеллектуальных способностей человека, что неизбежно при злокачественной мышечной дистрофии (по Дюшену). Не столь существенны и изменения в мышечной ткани сердца, поэтому при рассматриваемой болезни практически не наблюдается кардиомиопатия, либо она встречается в легкой форме.
Как и при других формах мышечной дистрофии, клинический анализ крови показывает повышения уровня некоторых ферментов в сыворотке крови, хотя они и не такие значительные как в случае с изменениями по Дюшену. Происходят сбои и в обменных процессах
[16], [17], [18], [19], [20], [21], [22], [23]
Данную патологию называют еще юношеской. Симптомы этого заболевания начинают появляться в период с десяти до двадцати лет. Существенным отличием симптоматики данной формы заболевания является то, что первичным местом локализации изменений является плечевой пояс, а уже далее атрофия мышц начинает захватывать все новые области организма больного: верхние конечности, затем область пояса, таза и ног.
Случаи заболевания встречаются в пропорции 15 больных на один миллион населения. Дефектный геном переходит наследственно, по аутосомно-рецессивному пути. Страдают от этого заболевания, с равной вероятностью, как женщины, так и мужчины.
Мышечная дистрофия эрба рота существенно деформирует грудную клетку больного (как бы проваливая ее назад), живот начинает выдаваться вперед, походка становится неуверенной, переваливающейся. Первые признаки заболевания появляются приблизительно в 14 – 16 лет, но сам диапазон гораздо шире: бывают случаи и более позднего развития - после третьего десятка, или наоборот – лет в десять (при ранней симптоматике болезнь протекает с более тяжелыми проявлениями). Интенсивность и развитие течения болезни от случая к случаю различно. Но средняя продолжительность цикла с момента появления первых симптомов до полной инвалидности составляет от 15 до 20 лет.
Чаще всего мышечная дистрофия эрба начинает проявляться с изменений в тазово-поясном отделе, а так же с отеков и слабости в ногах. Далее распространяющаяся патология постепенно захватывает и остальные мышечные группы организма больного. Преимущественно поражение не затрагивает мышцы лица, сердечная мышца остается не тронутой, уровень интеллекта, обычно, держится на прежнем уровне. Количественный показатель ферментов в сыворотке крови слегка увеличен, но не до такого уровня как в предыдущих случаях.
Мышечная дистрофия рассматриваемой формы является одной из самых аморфных патологий.
Рассматриваемое заболевание является наследственным и связанным с полом (дефект генома Х-хромосомы). Путь передачи – рецессивный.
Клиника проявления достаточно ранняя – до трехлетнего возраста малыша. Даже в грудничковый период можно заметить отставание в развитии моторики у карапуза, позже, чем здоровые детки, они начинают сидеть и ходить. Уже к трем годам у малыша, заметна слабость в мышцах, он быстро устает, плохо переносит даже незначительные нагрузки. Постепенно атрофия захватывает тазовый пояс и проксимальные мышцы нижних конечностей.
Медицина насчитывает ряд наследственных (генетических) заболеваний, поражающих мышечные и нервные ткани. Одна из них - нервно мышечная дистрофия, которая характеризуется нарушением моторных и статических проявлений на фоне мышечной атрофии. Поражению подвергаются нейроны, отвечающие за двигательные функции (клетки переднего рога), что приводит к изменениям группы тканей спинного мозга. Повреждение нейронов ядра клеток черепного нерва влияют на мимику лица, бульбарную и глазную мускулатуру. Так же за двигательные процессы отвечают однотипные клетки, при поражении которых страдают нервные окончания периферии и, нервно-мышечные соединения.
Базовые признаки такой патологии:
- Атрофия мышечно-соединительных тканей.
- Мышечные боли.
- Быстрая утомляемость больного.
- Снижение чувствительности рецепторов.
- Или наоборот повышенная чувствительность, вплоть до болевых синдромов.
- Появление внезапных судорог.
- Головокружения.
- Патология сердца.
- Ухудшение зрения.
- Сбой в системе потоотделения.
Чаще всего патология данной формы начинает проявляться у подростков в 10 – 15 лет, хотя фактически известны случаи, когда мышечная дистрофия ландузи дежерина начинала развиваться у шестилетних детей, либо у пятидесятилетнего человека. Первичной областью патологии, чаще всего, является группа мышц лицевой зоны. Постепенно ореол поражения расширяется, начинают атрофироваться группы плечевого пояса, торса и далее вниз. При поражении лицевой мимики в ранний период заболевания, неплотно закрываются веки. Приоткрытыми остаются и губы, что приводит к речевому дефекту. Течение болезни идет медленно – на протяжении этого периода человек абсолютно трудоспособен, только лет через 15 – 20 постепенно начинают атрофироваться мышцы пояса и таза – это и приводит уже к двигательной пассивности. И только к 40 – 60 годам поражение полностью захватывает нижние конечности.
То есть мышечную дистрофию ландузи дежерина можно назвать благоприятно текущим проявлением мышечного поражения.
Как и все предыдущие, мышечная дистрофия эмери дрейфуса является заболеванием наследственным. Основная зона поражения – атрофия плечелоктевых и голеностопных мышц. Данная болезнь характеризуется длительным периодом развития. В подавляющем большинстве случаев поражению подвергается сердце: брадиаритмияи, снижение кровяной проходимости, блокада и другие. Сбои в работе сердца могут быть причиной обмороков, а иногда даже летального исхода.
Ранняя диагностика не только самого заболевания, а и дифференцирование ее формы, поможет спасти жизнь не одному больному.
[24], [25], [26], [27], [28], [29]
Конечностно поясная мышечная дистрофия относится к наследственной патологии, путями наследования которой являются как аутосомно-рецессивные, так и аутосомно-доминантные болезни. Базовый район поражения – это область пояса, торса и верхних конечностей. При этом мышцы лицевой мускулатуры не страдают.
По данным исследований удалось установить как минимум два локуса генома хромосом, при мутации которых создается толчок к развитию конечностно поясной мышечной дистрофии. Прогрессирование данного поражения проходит достаточно медленно, давая больному в полной мере насладиться жизнью.
Аутосомно-доминантная болезнь, проявляющаяся уже в достаточно зрелом возрасте - окулофарингеальная мышечная дистрофия. Как не странно это звучит, но данная патология поражает людей, принадлежащих определенным этническим группам.
Чаще всего симптоматика начинает проявляться к 25 – 30 годам. Классическими признаками данной мышечной дистрофии является атрофия лицевых мышц: птоз век, проблемы с глотательной функцией (дисфагия). Болезнь, постепенно прогрессируя, приводит к неподвижности глазного яблока, при этом внутренние мышцы глаза не подвергаются поражению. На этом этапе изменения могут остановиться, но иногда патологии подвергаются и остальные лицевые мышцы. Достаточно редко, но бывают задействованы в разрушительном процессе и группы мышц плечевого пояса, шеи, неба и глотки. В этом случае кроме офтальмоплегии и дисфагии прогрессирует еще и дисфония (проблема речевого аппарата).
Мышечная дистрофия у детей
Детство. Многие его вспоминают с улыбкой. Прятки, качели, велосипеды… Да сколько еще различных игр придумывает детвора. Но есть малыши, которые не могут себе позволить такой роскоши. Мышечная дистрофия у детей не дает такой возможности.
Практически все, за редким исключением, формы могут проявляться у деток своей симптоматикой: и злокачественная форма патологии по Дюшону (развивающаяся только у мальчиков), и доброкачественная мышечная дистрофия по Беккеру и другие. Особенно опасна патология, развивающаяся стремительно, агрессивно (форма по Дюшону). Причем для малыша опасна даже не столько сама симптоматика (атрофия практически всех групп мышц), как вторичные осложнения, которые и приводят к двадцати годам к смерти. Чаще всего летальный исход наступает вследствие респираторной инфекции или сердечной недостаточности. Но данная симптоматика становится более явной только тогда, когда малыш начинает делать первые шаги.
- Задержка в развитии: такие дети позже начинают сидеть и ходить.
- Медленное интеллектуальное развитие.
- Первыми поражаются мышцы позвоночника.
- Таким малышам трудно бегать и подниматься по лестнице.
- Походка вперевалочку.
- Деформация позвоночника.
- Ходьба на пальцах.
- Малышу тяжело держать свой вес, и он быстро утомляется.
- За счет жировой ткани увеличивается размер мышц.
- Поражение конечностей идет симметрично.
- Патологическое увеличение челюсти и промежутков между зубками.
- Приблизительно с 13 лет малыш перестает ходить совсем.
- Патология сердечной мышцы.
При других формах поражения, симптоматика достаточно похожа, только тяжесть поражения значительно ниже.
Читайте также:
