
Мышечно глазо мозговая болезнь
| МКБ-10 | Q04.3 |
|---|---|
| МКБ-9 | 742.2 |
| OMIM | 236670 247200, 253280, 253800, 257320, 300121, 300215, 304800, 601545, 611603 |
| DiseasesDB | 29492 |
| MeSH | D054082 |
Содержание
- Общие сведения
- Формы
- Причины развития
- Патогенез
- Симптомы
- Диагностика
- Лечение
Возникает вследствие недостаточной миграции зародышевых нервных клеток (нейробластов) из первичной нервной трубки.
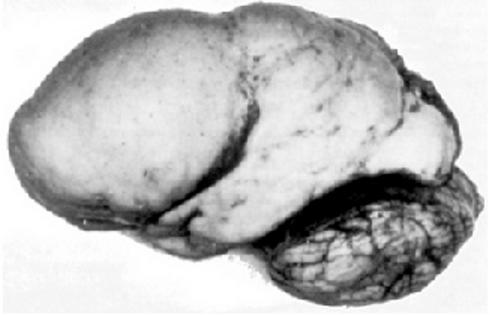
Наиболее тяжелая форма заболевания – агирия, при которой извилины мозга вообще отсутствуют.
Общие сведения
Лиссэнцефалия – редко встречающийся порок развития, который может быть как самостоятельной аномалией развития, так и проявлением некоторых врожденных синдромов (Миллера-Дикера и Нормана-Робертса). Наблюдается у детей независимо от пола. Распространенность заболевания в настоящее время не установлена.
Формы
Лиссэнцефалия может быть тотальной и очаговой.
В зависимости от тяжести нарушений выделяют:
- агирию, при которой поверхность мозга лишена извилин;
- пахигирию, которая отличается наличием нескольких плоских широких извилин и неглубоких борозд.
В зависимости от морфологических и генетических данных с 2003 года выделяется пять групп данной аномалии.
Первая группа — классический тип заболевания (I тип лиссэнцефалии в предшествующей классификации), которая включает:
- лиссэнцефалию, связанную с мутацией гена LIS1;
- лиссэнцефалию, которая вызывается мутацией гена DCX (doublecortin);
- изолированную лиссэнцефалию I типа;
- синдром Миллера-Дикера;
- изолированную лиссэнцефалию I типа, которая возникла по неустановленным генетическим причинам.
Во вторую группу включена форма, которая связана с X-сцепленным рецессивным наследованием и сопровождается врожденным отсутствием мозолистого тела. Заболевание вызывается мутацией гена ARX.
Третья группа включает лиссэнцефалию, при которой наблюдается врожденное недоразвитие (гипоплазия мозжечка). К этой же группе относится синдром Норман-Робертс, вызванный мутацией гена RELN.
К четвертой группе относят микролиссэнцефалию.
- синдром Уокера-Варбурга (HARD(E)-синдром);
- синдром Фукуямы;
- Мышечно-Глазо-Мозговую Болезнь (MEB).
Существует также морфологическая классификация, выделяющая:
Причины развития
Лиссэнцефалия развивается в результате нарушения миграции нейронов при формировании и развитии коры головного мозга.
Нарушение миграции возможно в результате мутации:
- гена RELN (рилина), который расположен на хромосоме 7 в регионе 7q22 (развивается синдром Норман-Робертс);
- гена ARX, расположенного на Х-хромосоме в регионе p21.3.
К нарушениям формирования коры головного мозга приводит и частичная утрата (делеция) гена LIS-1, расположенного в хромосоме 17 регион р13.3 и принимающего участие в миграции нейронов (аутосомно-рецессивный тип наследования).
При утрате некоторых генов 17-й хромосомы в регионе 13 (PAFAH1B1) развивается синдром Миллера-Дикера.
К развитию лиссэнцефалии приводят также нарушения X-сцепленных генов. При мутациях гена XLIS у детей мужского пола диагностируют лиссэнцефалию, а у детей женского пола – двойную кору, так как в результате нарушения миграции слой серого вещества откладывается под белым веществом головного мозга (в норме должно быть наоборот). Дефектный ген кодируется белком doublecortin (DCX), поэтому его мутация тоже может стать причиной развития аномалии. Тип наследования — Х-сцепленный рецессивный.
- Дефекты 9-й хромосомы в регионе q34 (синдром Уокера-Варбурга).
- Дефекты гена FCMD, расположенного в 9-й хромосоме в регионе q31 (синдром Фукуямы). Чаще всего мутация заключается в присутствии избыточных мобильных генетических элементов первого типа.
- Мутации гена POMGNT1, который расположен в 1-й хромосоме в регионе p34 (Мышечно-Глазо-Мозговая Болезнь).
Причины некоторых форм лиссэнцефалии в настоящее время не выявлены.
Кроме генетических нарушений причиной развития аномалии может быть:
- перенесенная в первом триместре вирусная инфекция матки или плода;
- плохое кровоснабжение мозга плода на начальном этапе беременности.
Патогенез
Формирование и развитие коры головного мозга включает:
- пролиферацию (деление) нервных клеток;
- миграцию нейронов из эмбрионального матрикса;
- дифференцировку коры.
При нормальном развитии плода происходит:
- оформление полушарий мозга на 11-12 неделе внутриутробного развития;
- формирование латеральной, шпорной и опоясывающей борозды на 16 неделе эмбриогенеза;
- уплощение кортикального плато и формирование первичных и центральных борозд на 20 неделе;
- формирование ольфакторных борозд на 24-26 неделе;
- образование верхних височных борозд на 28 неделе.
Созревание коры головного мозга обеспечивается миграцией нейронов, которые располагаются в мозге слоями. В процессе миграции те клетки, которые образовались позже, мигрируют дальше своих предшественниц, образуя таким образом новый слой.
Процесс миграции регулируется белками рилином и N-кадгерином.
Большая часть борозд и извилин формируются после окончания миграции нейронов, поэтому нарушение процесса миграции нейронов приводит к нарушению формирования извилин и борозд.
Симптомы
Симптомы заболевания зависят от типа лиссэнцефалии.
Классический тип заболевания проявляется:
Лиссэнцефалия при синдроме Миллера-Дикера сопровождается:
- черепно-лицевой дизморфией (изменением формы лица);
- увеличенным расстоянием между парными органами;
- замедленным ростом;
- патологиями почек, сердца, ЖКТ.
При обследовании выявляется уменьшенное количество кортикальных слоев (четыре вместо шести).
При синдроме Нормана-Робертса заболевание сопровождается:
- увеличенным расстоянием между глазами;
- микроцефалией (маленький размер головы);
- наличием покатого лба;
- эпилепсией;
- отеком тканей (лимфедемой);
- задержкой умственного развития.
Обследование позволяет выявить утолщенную кору мозга, наличие агирии, значительные изменения гиппокампа, мозжечка и ствола мозга.
При синдроме Уокера-Варбурга помимо лиссэнцефалии наблюдается:
- микрофтальмия, катаракта, отслоение сетчатки;
- аномалии развития скелета и мышц;
- гидроцефалия, цефалоцеле;
- задержка умственного развития.
При синдроме Фукуямы лиссэнцефалия сочетается с:
- прогрессирующей мышечной дистрофией;
- задержкой темпов развития;
- выраженным слабоумием;
- гидроцефалией, микрополигирией, аплазией мозолистого тела, уменьшением плотности белого вещества;
- судорогами в большинстве случаев.
В отдельных случаях выявляют атрофию зрительных нервов и патологию сетчатки.
Диагностика
Для диагностики лиссэнцефалии используются:
- пренатальное обследование на 22- 28 неделе беременности при помощи ультразвукового оборудования;
- КТ, позволяющая выявить наличие сглаженности извилин, гипоплазию белого вещества, увеличение желудочков мозга, прямолинейную границу между серым и белым веществом, расширение и вертикальную ориентацию сильвиевых щелей, расширение центральной борозды и др.;
- МРТ, которая позволяет выявить тип лиссэнцефалии по характерным признакам;
- ЭЭГ, при помощи которого определяется характерный для лиссэнцефалии быстрый и высокоамплитудный ритм в задних отделах мозга;
- генетический анализ.
При пренатальной диагностике необходимо учитывать, что в начале II-го триместра достаточно гладкий мозг – норма, поэтому оценивать формирование извилин и борозд можно не раньше 20 недели.
При подозрении на наличие аномалии возможно исследование хромосом плода.
Лечение
Специфическое лечение лиссэнцефалии не разработано.
Для улучшения состояния больных применяются:
- производные вальпроевой кислоты в качестве противосудорожной терапии;
- карнитин, который используется при мышечной дистрофии;
- церебрализин, актовегин, глицин, витамины в качестве поддерживающей терапии.
В некоторых случаях возможно оперативное вмешательство, которое заключается в трансплантации стволовых клеток. Поскольку положительные результаты пока наблюдаются редко и заключаются только в некотором улучшении состояния, а риск побочных эффектов у больного ребенка высок, операции проводят не часто.
Лиссэнцефалия – заболевание с неблагоприятным прогнозом, поэтому семьям, в которых имелось данное заболевание, необходима консультация генетика.
Автор - д.м.н. Елена Леонидовна Дадали Письмо Автору
ВРОЖДЕННАЯ МЫШЕЧНАЯ ДИСТРОФИЯ С РИГИДНОСТЬЮ ПОЗВОНОЧНИКА 2 ТИПА, МЕРОЗИН-ПОЗИТИВНАЯ (OMIM: 602771)
Это заболевание из группы синдромов ригидного позвоночника, впервые описанного Dubowitz в 1973 году.
Заболевание является классическим вариантом врожденной непрогрессирующей миопатии, характеризующейся слабостью и гипотрофиями мышц туловища и конечностей, контрактурами и дистрофическими изменениями в биоптате мышц. Наряду с мышечно-глазо- мозговым синдромом, склероатонической миодистрофией Ульриха и синдромом Уолкера-Варбурга эта нозологическая форма составляет группу мерозин-позитивных врожденных миопатий. Эта группа заболеваний составляет 50% всех случаев врожденных структурных миопатий.
Первые признаки заболевания возникают с рождения или в периоде новорожденности и характеризуются диффузной мышечной гипотонией с преимущественным поражением проксимальных групп мышц, лицевой, глазодвигательной и дыхательной мускулатуры и контрактурами суставов и сколиозом. У большинства больных отмечаются трудности вскармливания, эпизоды апноэ, респираторный дистресс-синдром. Несмотря на раннее возникновение клинических проявлений большинство 92% больных не имеют выраженной задержки темпов моторного развития и сохраняют способность к самостоятельной ходьбе до 4-х летнего возраста. Характерной особенностью этого заболевания является ограничение разгибания в шейном и поясничном отделах позвоночника, а также крупных суставах конечностей, деформация позвоночника и грудной клетки, часто приводящее к возникновению дыхательных расстройств.
Для этой формы заболевания в отличии от мерозин-негативной врожденной мышечной дистрофии не характерно поражение белого вещества мозга. Признаки поражения белого вещества имеют только 11% больных.
При электромиографическом обследовании выявляется первично-мышечный характер поражения.
В ряде случаев отмечается нерезко выраженное повышение активности креатинфосфокиназы.
Специфичных морфологических дефектов не выявлено. В биоптате мышечных волокон выявляются неспецифические изменения общие для первично-мышечного характера поражения.
Предполагается, что тип наследования заболевания аутосомно-рецессивный с варьирующей пенетрантностью. Учитывая значительное преобладание больных мужского пола не исключена ограниченная полом экспрессивность гена.
Предполагается наличие генетической гетерогенности заболевания.
Показано, что одна из нозологических форм обусловлена мутациями в гене ламинина альфа-2 цепи (мерозина) локализованного на хромосоме 6q, в другой семье ген заболевания картирован в хромосомном регионе 1р36-р35, Один из генов заболевания, мерозин (LAMA2, laminin M, merosin, OMIM: 156225) картирован в области 6q22-q23 и содержит 64 экзона. Размер гена более 260 тысяч п.н.. Два из его экзонов чрезвычайно малы – их размер равен соответственно 6 и 12 п.н.. Другой ген SEPN1 (SELENOPROTEIN N 1,SELN, OMIM: 606210) был картирован Moghadaszadeh и сотр., в 2001 году. SEPN1 включает 13 экзонов и имеет длину более 18,5 тысяч п.н.. Не исключается наличие дальнейшей генетической гетерогенности, а также участия нескольких генов в возникновении одной и той же нозологической формы.
- Dubowitz, V.: Rigid spine syndrome: a muscle syndrome in search of a name. Proc. Roy. Soc. Med. 66: 219-220, 1973.
- Moghadaszadeh, B.; Desguerre, I.; Topaloglu, H.; Muntoni, F.; Pavek, S.; Se- wry, C.; Mayer, M.; Fardeau, M.; Tome, F. M. S.; Guicheney, P.: Identification of a new locus for a peculiar form of congenital muscular dystrophy with early rigid- ity of the spine, on chromosome 1p35-36. Am. J. Hum. Genet. 62: 1439-1445, 1998.
- Moghadaszadeh, B.; Petit, N.; Jaillard, C.; Brockington, M.; Roy, S. Q.; Mer- lini, L.; Romero, N.; Estournet, B.; Desguerre, I.; Chaigne, D.; Muntoni, F.; Topa- loglu, H.; Guicheney, P.: Mutations in SEPN1 cause congenital muscular dystro- phy with spinal rigidity and restrictive respiratory syndrome. Nature Genet. 29: 17- 18, 2001.
В итоге поверхность мозга становится удивительно гладкой. Этот дефект начинает проявляться еще с 12 недели развития плода, и заканчивает формироваться спустя 2 недели, и вызван он нарушенным процессом передвижения нейронов в этот период.
Головной мозг, в нормальном состоянии у каждого здорового человека, имеет множество складок и борозд. У плода с этим отклонением, структура развивается не своеобразным способом, у таких пациентов складки и борозды либо частично отсутствуют, либо их вовсе нет, и произошедшие изменения уже не обратимы.
Точно так же, как и многие другие дефекты в развитии головного мозга, лиссэнцефалия берет под контроль большой спектр фенотипов, они могут колебаться в соответствии с тяжестью от переменной агирии, либо до полной.
Сопутствующие заболевания и нарушения
Сопутствовать данному пороку развития может — синдром Миллера-Дикера, либо Уокера-Варбурга.
Синдром Миллера-Дикера характеризуется классической лиссэнцефалией, при таком синдроме под воздействие аномалии попадают лицевые

мышцы и проявляются другие возможные дефекты, которые можно встретить у людей только с таким синдромом.
Особенностью синдрома является потеря в хромосоме 17 нескольких генов. За лиссэнцефалию отвечает потеря гена PAFAH1B1, это доказанный учеными факт.
А вот при утере гена YWHAE, лиссэнцефалия может усложниться. Последствия после потери других генов при данном синдроме еще не известны.
Синдром Уокера-Варбурга – очень редкая врожденная дистрофия мышц, связанная с нарушением работы головного мозга и лицевых мышц.
Классификация заболевания
Развитие патологии происходит из-за мутировавшего гена PAFAH1B1. Такой дефект можно разделить на изолированную лиссэнцефалию и синдром Миллера-Дикера.
Изолированная патология проходит без развития других дефектов в геноме, во втором случае наблюдается развитие сопутствующих аномалий из-за мутации в DCX гене.
- заболевание – мышцы-глаз-мозг;
- синдромы Уокера-Варбурга и Фукуямы.
Кроме этого, существуют также другие подтипы:
- микролиссэнцефалия;
- синдром Нормара-Робертса;
- патология вызванная мутировавшим ARX геномом.

Типы развития
Рассмотрим патологию, базируясь на 4 типах развития.
Мозг ребенка можно сравнить с мозгом плода на 23 неделе развития. Кора состоит из четырех слоев нейронов, развивается пахигирия, агирия.
- гладкая наружная часть коры мозга;
- сильно уменьшен объем белого вещества в мозгу;
- ленточная гетеротопия, которая отделена от коры белой полосой;
- гипоплазия ствола.
Кора мозга значительно толще, чем должна быть, без нормального слоевого строения, имеются нарушения в сосудистом древе; присутствует 
гидроцефалия.
- гладкая поверхность коры;
- гипоплазия;
- размыта граница между белым и серым веществом;
- утолщение коры;
- агенезия мозолистого тела;
- у некоторых больных в затылочной области – энцефалоцеле.
- белое вещество в меньших количествах, нежели нужно;
- гипоплазия;
- начинают выделяться извилины, но это малозаметно.
- остановка миелинизации;
- головной мозг очень мал в размере;
- количество нейронов составляет 35% нормы;
- на коре виднеются некоторые извилины и борозды с нормальной толщиной.
В некоторых случаях используют еще одну классификацию лиссэнцефалии, ее основывают в зависимости от тяжести болезни:
- самый тяжелый класс, тотальная агирия, нет никаких извилин;
- имеется небольшое количество складок на затылочных и лобных полосках, диффузная агирия;
- сочетание пахигирии и агирии;
- диффузная пахигирия;
- пахигирия спереди и гетеротопия;
- гетеротопия под корой.
Первая и четвертая степень очень редка, вторая имеется у детей с синдромом Миллера-Дикера. Самое частое — сочетание в третьей степени, обычно такая патология состоит из задней агирии и лобной пахигирии.
Тип первый, то есть классический, встречается у 12 человек на миллион рожденных.
Причины и патогенез аномального развития

Определить из-за чего начал формироваться дефект — задача не из легких, так как их причин быть целое множество. Примерно у 60% людей с лиссэнцефалией замечены мутация в гене LIS1.
Еще у примерно 3% людей, конкретно с классическим типом патологии, можно отметить мутацию в гене TUBA1A. Примерно у 30% пациентов с гипоплазией наблюдаются изменения в TUBA1A гене.
Иные причины возникновения могут быть самыми разными, например, при развитии инфекционных заболеваний в матке в первом триместре беременности, или же на ранних сроках у плода было плохое кровоснабжение в области мозга, в результате чего начала развиваться лиссэнцефалия.
Процесс развития коры головного мозга:
- нервные клетки начинают делиться;
- кора дифференцируется;
- нейроны движутся из матрикса.
Хронология развития плода в норме должна происходить таким образом:
- на 11-12 неделях начинают формироваться оба полушария;
- на 16 – начинают формироваться борозды и извилины;
- на 20 — уплотняется кортикальное плато, начинают формироваться первичные и центральные борозды;
- на 23-26 — сформировываются ольфакторные борозды;
- на 28 неделе развития внутри утробы матери, начинают формирование борозды в височной области.
Кора головного мозга формируется за счет миграции мозговых клеток – нейронов, которые находятся в разных слоях мозга. В процессе их передвижения, клетки, которые сформировались на более позднем этапе, передвигаются дальше, чем предыдущие, в результате происходит формирование нового слоя. Этот процессе регулируется специальными белками N-кадгерином и рилоном.
В норме, все основные борозды и извилины начинают свое формирование после окончания передвижения нейронов, именно нарушение данного процесса влияет на неправильное создание извилин и борозд.
Постановка диагноза и возможности медицины
Обычно в таком случае диагноз ставится при рождении или немного позже, после проведения УЗИ и исследования результатов КТ или МРТ.
Но врачи могут заподозрить наличие патологии еще во время беременности, ведь они регулярно проводят УЗИ плода, и постоянно изучают его состояние, в частности и развитие головного мозга.
Если врач заподозрит лиссэнцефалию, кроме УЗИ нужно будет проводить еще несколько методов диагностики, например, анализ на явность мутирующих генов и ЯРМ-томографию.
Обнаружить данную патологию можно не раньше, чем на 20 неделе беременности, поскольку поверхность мозга до этого времени, и без каких-либо нарушений, является гладкой. Именно после данного периода, можно заметить отклонения в развитии плода.
Вылечить данный порок невозможно, есть возможность симптоматического лечения, и оно зависит от стадии развития и места расположения дефектов. Необходимо поддерживать постоянный уход.
Для людей с гидроцефалией обычно делают шунтирование, а для контроля судорог принимаются специальные лекарственные препараты.
Прогноз продолжительности жизни
Главным фактором, который определяет прогноз продолжительности жизни ребенка с диагнозом лиссэнцефалия, является степень тяжести заболевания. Так некоторые из пациентов имеют более-менее нормальное интеллектуальное развитие.
В основном дети, с более тяжелой формой патологии, не способны прожить дольше 10 лет, но даже дожив до этого возраста, их развитие все равно остается на уровне 4-6 месячного ребенка.
Главной причиной смерти больных, в основном, является связь с аспирацией питательных продуктов или жидкости, с очень тяжелыми приступами, или с болезнями дыхательных путей.
Врожденные миопатии — это широкое понятие нервных мышечных расстройств, группа редких первичных дефектов мышц, которые передаются по наследству, вызывают гипотонию при рождении или в период новорожденности. Заболевания, относящиеся к группе врожденных миопатий, имеют сложную клиническую картину и схожие симптомы, что значительно усложняет их диагностирование и лечение.
- Классификация врожденной миопатии
- Симптомы врожденной миопатии
- Характеристика отдельных видов врожденной миопатии
- Диагностика врожденной миопатии
- Лечение врожденной миопатии
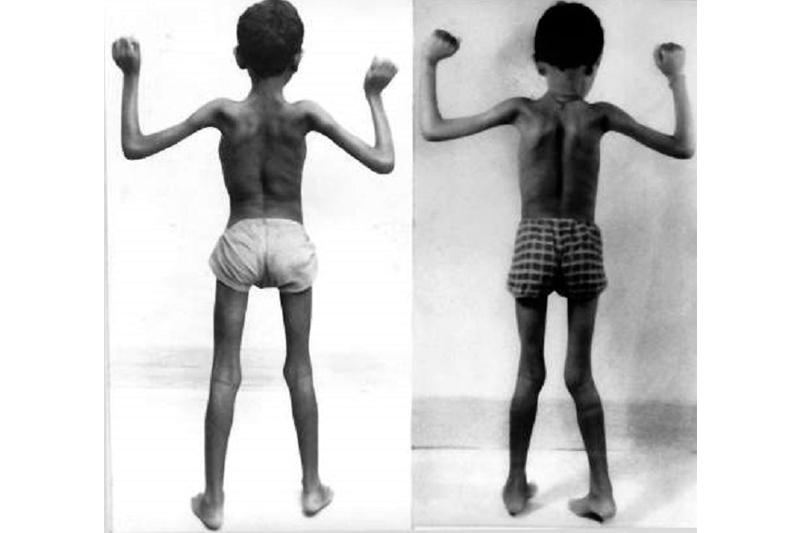
Проявляется как диффузная мышечная слабость и снижение мышечного тонуса, степень выраженности которых напрямую зависит от разновидности миопатии и уровня ее сложности. В тяжелых случаях может привести к летальному исходу от дыхательной недостаточности.
Врождённая миопатия — заболевание генетически обусловленное. Различные виды миопатии могут находиться в разных локусах хромосом, поэтому могут передаваться по наследству рецессивно, доминантно или совместно с Х-хромосомой. Генетическая патология нарушает синтез белка, который входит в состав мышечной ткани, что приводит к нарушению строения мышечных волокон. В следствие этого, мышцы утрачивают возможность нормально сокращаться, наблюдается мышечная слабость.
Проявляется врожденная миопатия обычно в раннем детском возрасте (очень редко проявляется у взрослых) и сохраняет свою симптоматику на протяжении всей жизни больного. Чаще всегоданное заболевание слабо прогрессирует или не прогрессирует совсем.
Классификация врожденной миопатии
Биопсия мышц дает определенные биохимические и морфологические данные, на основании которых все заболевания врожденной миопатии можно условно разделить две большие группы.
Врожденные мышечные дистрофии
Первая группа — это врожденные мышечные дистрофии (нарушение функции и строения мышечных волокон в результате сбоя синтеза белков, которые входят в их состав). Четкой классификации врожденных мышечных дистрофий нет, но на основании гипотез патогенеза данной группы заболевания можно выделить две группы:
- мерозин-негативные заболевания, которые характеризуются дефицитом или отсутствием белка мезонина, входящего в состав поперечно-полосатых мышц;
- врожденные структурные миопатии и мерозин-позитивные, при которых концентрация мезонина находится в норме.
Мерозин-негативная группа заболеваний подразделяется на несколько типов:
- врожденные мышечные дистрофии Фукуямы;
- мышечно-глазо-мозговой синдром;
- синдром Уолкера-Варбурга.
Клиническая картина заболеваний мерозин-негативной группы очень схожа с симптоматикой классических врожденных миопатий, но отличительной особенностью является вовлечение в общую симптоматику различных структур головного мозга, что ведет к дальнейшей умственной отсталости, задержке развития. Заболевания мерозин-позитивной группы намного реже включают в себя поражение центральной нервной системы (приблизительно у 10% больных выявлено поражение мозга) и обычно не влечет за собой торможение интеллекта. Клиническая картина характеризуется деформацией позвоночника и нарушением черт строения лица.
Врожденные структурные миопатии
Вторая группа — врожденные структурные миопатии (нарушения целостности цитоскелета мышечных волокон и возникновение патологии в биоптате мышц). Эта группа заболеваний характеризуется нарушением синтеза белков, отвечающих за рост и другие функции формирования мышц в раннем развитии эмбриона.
К врожденным структурным миопатиям относят:
- болезнь центрального стержня;
- немалиновую миопатию;
- центронуклеарную миопатию;
- мегакониальную миопатию;
- миопатию с диспропорцией типов мышечных волокон;
- миопатию со множественными центральными стержнями;
- миотубулярную миопатию;
- миопатию с кристаллическими включениями.
Клинические картины каждого из заболеваний данной группы похожи друг на друга и характеризуются мышечной гипотонией и гипертрофией, пониженной рефлективностью в сухожильях и повышением концентрации в крови креатинфосфокиназы. Наблюдается медленное прогрессирование.
Симптомы врожденной миопатии
Слабость в мышцах может быть выражена сильно или незначительно. Чаще всего симптоматика сохраняется на весь период жизни больного и практически не прогрессирует или слабо развивается. В отдельных случаях, можно наблюдать невозможность самостоятельно передвигаться, поэтому больной вынужден использовать коляску, но навыки самообслуживания, приобретенные им, не утрачиваются.
Врожденные миопатии провоцируют не только слабость мышц конечностей и спины, слабеют и мышцы дыхательной мускулатуры, что является особенно опасным для детей грудного возраста. Если мышечная слабость дыхательных путей выражена в малой степени, то наблюдается развитие дыхательной недостаточности. Это, в свою очередь, провоцирует различные заболевания дыхательных путей (бронхиты, всевозможные виды пневмоний). Иногда дыхательная недостаточность приводит к летальному исходу еще в младенческом возрасте. Бывают случаи, когда с возрастом слабость мышц уменьшается или наоборот прогрессирует.
В отдельных случаях врожденная миопатия проявляется также в виде дисморфичных черт лица (удлиненная форма черепа, высокое небо) или патологиями развития скелета (сколиоз, косолапость, врожденный вывих бедра, кифоз).
Характеристика отдельных видов врожденной миопатии
Болезнь центрального стержня
Наследуется аутосомно-доминантным способом с неполной пенетрантностью (но встречаются и спорадические случаи наследственности). Данная форма врожденной миопатии характеризуется патологией проксимальных мышц конечностей, но больные способны приобрести некоторые двигательные навыки. В младенческом возрасте наблюдается задержка двигательного развития и гипотония, но диагностировать данное заболевание можно только в более позднем возрасте при изменениях скелета и выраженной мышечной слабости. При этом наблюдаются патологии скелета: деформация стоп, кифосколиоз, дислокация бедер, грудь сапожника.
Чаще всего, больные имеют хрупкую фигуру и невысокий рост. При диагностике заболевания проводят биоптат мышц, который показывает наличие множественных или единичных прерывистых зон, которые лишены ферментов окисления, в некоторых мышечных волокнах. Проведение других лабораторных анализов может показать норму. Пациенты с болезнью центрального стержня склонны к развитию злокачественной гипертермии.
Немалиновая миопатия
Второе название данного заболевания — врожденная непрогрессирующая нитеобразная миопатия. Наследственность в основном передается по аутосомно-доминантному типу, но встречается также рецессивный и спорадический. Возможен летальный исход вследствие дыхательной недостаточности в раннем младенческом возрасте. Наблюдается сильно выраженные патологии скелета. Развитие болезни может происходить в той или иной степени, а может не прогрессировать вовсе. В отдельных случаях больные вынуждены передвигаться с помощью сидячей каталки, в других — страдают от дыхательной недостаточности. При диагностировании проводится гистологическое исследование, которое выявляет в мышцах неподобные или палочкоподобныене малиновые тельца. ЭМГ обычно утверждает диагноз миопатии.
Миотубулярная миопатия
Данный тип врожденной миопатии наследуется по аутосомно-рецессивному типу. Миотубулярная миопатия характерна наличием центрально расположенных ядер в большинстве мышечных волокон. Это напоминает вид мышцы на миотубулярном внутриутробном развитии плода. В следствии этого заболевание и получило свое название.
Диагностика врожденной миопатии
Диагностика при врожденных миопатиях — сложный процесс, поскольку врачу необходимо дифференцировать и определить конкретный вид миопатии для назначения адекватного лечения и постановки правильного диагноза. Врач-невропатолог выявляет неврологические симптомы, проводит электрофизиологическое и биохимическое исследование, чтобы установить гетерозиготное носительство миопатического гена. Электромиографическое исследование с помощью накожных электродов показывает зачастую снижение вольтажа кривой ЭМГ. При биохимическом анализе крови в сыворотке наблюдается повышенная концентрация альдолазы и креатинкиназы.
Лечение врожденной миопатии
Лечение врожденный миопатий малоэффективное. Четкого лечения на данный момент нет. О том, можно ли лечить врожденную миопатию, ученые спорят до сих пор. Медицинские институты разных стран проводят исследования на генном уровне — с использованием стволовых клеток. Существует симптоматическое лечение, которое заключается в воздействии на обменные процессы в организме пациента, в частности, синтез белков, попытку нормализации функций вегетативной нервной системы. Чаще всего медикаментозное лечение включает в себя принятие анаболических гормонов (неробол, цераксон, ретаболил, сомазин), АТФ. Обязательно проводится витаминотерапия. Назначаются также антихолинэстеразные препараты.
Обязательным звеном процесса лечения врожденных миопатий является лечебная физкультура. Это могут быть занятия в воде или комплекс упражнений. ЛФК контролируется тренером или неврологом. В отдельных случаях действенной оказывается ортопедическая коррекция (например, ношение ортопедической обуви, корсетов или использование ортопедических матрасов, подушек, кресел).
Состояние и клиническую картину заболевания контролирует невропатолог, терапевт, педиатр, кардиолог и ортопед-травматолог.
Читайте также:
