
Миопатия или мышечная дистрофия лечение

Мышечная дистрофия (МД) — это группа заболеваний, характеризующихся прогрессирующей слабостью и мышечной дегенерацией. Мышцы постепенно атрофируются — теряют свой объем и, следовательно, силу.
Это болезни генетического происхождения, которые могут возникать в любом возрасте: с самого рождения, в детстве или во взрослой жизни. Существует более 30 форм заболеваний, которые различаются по возрасту появления симптомов, характеру пораженных мышц и степени тяжести. Большинство типов дистрофий постепенно осложняются и имеют необратимые последствия. В настоящее время лечения МД все еще не существует. Наиболее известным и распространенным типом заболеваний является миопатия Дюшенна.
В ходе развития МД страдают в первую очередь основные мышцы, которые способствуют произвольному движению, включая мышцы, бедра, ног, рук и предплечья. В некоторых случаях могут быть затронуты респираторные мышцы и сердце. Люди с мускульной дистрофией постепенно теряют свою мобильность при ходьбе. Другие симптомы могут быть связаны с мышечной слабостью, включая сердце, желудочно-кишечные, глазные проблемы.
Распространенность заболевания
Миопатия относится к редким и неизлечимым заболеваниям. Трудно вывести точную статистику, поскольку она объединяет различные болезни. Согласно некоторым исследованиям, около 1 из 3 500 человек страдают от этого заболевания.
Например:
![]()
Миопатия Дюшенна затрагивает приблизительно одного ребенка (мальчика) из 3500.- Миопатия Беккера касается 1 мальчика из 18 000.
- Фазио-скапулогумаральная дистрофия поражает около 1 из 20 000 взрослых людей.
- Болезнь Эмери-Дрейфус затрагивает 1 из 300 000 человек, вызывает ретракцию сухожилия и нарушение сердечной мышцы
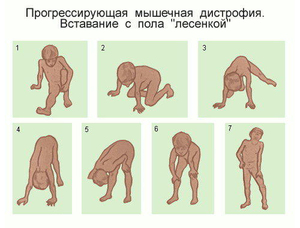
Частота и тип заболеваний зависит от конкретной страны:
Причины заболевания и лечение
Причиной данной патологии являются генетические заболевания, то есть дефект (или мутация) гена, необходимого для нормального развития мышц. Когда этот ген мутирует, мышцы больше не в состоянии нормально функционировать — они теряют свой силовой потенциал и в результате атрофируются.
Например:
- Миопатия Дюшенна связана с дефицитом дистрофина — белка, расположенного под мембраной мышечных клеток, который играет роль в сокращении мышц.
- Почти у половины врожденных МД причиной является дефицит мерозина — белка, составляющего мембрану клеток мышцы.
Как правило, МД передается рецессивно. Другими словами, для того чтобы болезнь выражалась, оба родителя должны быть носителями и передавать ребенку ненормальный ген. Болезнь не проявляется у родителей по той причине, что у каждого из них есть только один аномальный ген, а не два. Для нормального функционирования мышц достаточно одного нормального гена.
Кроме того, некоторые формы миопатии затрагивают только мальчиков: это миопатия Дюшенна и Беккера. В обоих случаях ген, участвующий в этих двух заболеваниях, расположен в Х-хромосоме, которая существует в единственной копии у мужского пола.
Симптомы заболевания
МД проявляются мышечной слабостью, которая имеет тенденцию к постепенному ухудшению, симптомы варьируются в зависимости от типа патологии. В зависимости от случая могут присутствовать и другие симптомы, такие как сердечные и респираторные расстройства, аномалии глаз (пороки развития, катаракта), интеллектуальный дефицит, гормональные нарушения и т. д.
Дети часто жалуются на судороги и мышечные боли. Болезнь развивается довольно быстро, как только появляются первые симптомы. Обычно использование инвалидной коляски требуется примерно в возрасте 12 лет. Такого рода нарушения приводят к сколиозу и деформациям суставов. Кроме того, у некоторых детей наблюдается умственная отсталость. К концу подросткового возраста часто возникают сердечные осложнения (сердечная недостаточность), а также респираторные проблемы, требующие искусственной подачи воздуха. Средняя продолжительность жизни (от 20 до 30 лет в среднем).
Миопатия Беккера. Симптомы сравнимы с симптомами М. Д. Дюшенна , однако они менее выражены, а развитие заболевания происходит медленнее. Симптомы начинаются в 5−15 лет, иногда позже, характеризуются прогрессирующей потерей силы мышц в конечностях и в окрестностях туловища. В более чем половине случаев ходьба остается возможной до возраста 40 лет.
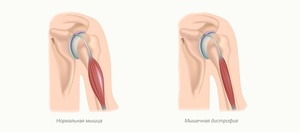
Миопатия Штейнтера. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Также наблюдается Миотония — аномальное и продолжительное сокращение мышц (мышца расслабляется слишком медленно), особенно выражается в руках, а иногда и на языке. Также могут быть затронуты мышцы лица, шеи и лодыжек. Часто присутствуют сердечные и дыхательные нарушения, которые являются потенциально серьезными. Нередко наблюдаются пищеварительные, гормональные, глазные расстройства, а также бесплодие и раннее облысение.
Миопатия поясничного отдела. Симптомы обычно проявляются в детстве (10 лет) или в раннем взрослом возрасте (около 20 лет). Мышцы плеч и бедер постепенно ослабевают, в то время как мышцы головы, шеи и диафрагмы обычно не затрагиваются. Если некоторые формы сопровождаются дыхательными нарушениями, то при этом типе дистрофии такие аномалии отсутствуют. Сердечные нарушения встречаются редко. Эволюция (развитие заболевания) очень изменчива, в зависимости от формы.
Миопатия Дежерина-Ландузи или плечелопаточная дистрофия. Симптомы обычно появляются в позднем детстве или в зрелом возрасте (от 10 до 40 лет). Как следует из названия, миопатия затрагивает мышцы лица, плеч и рук. Таким образом, больному становится сложно выразить улыбку, произнести некоторые предложения и закрыть глаза. Потеря подвижности происходит примерно в 20% случаев. Заболевание развивается медленно, продолжительность жизни нормальная.
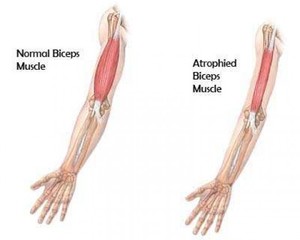
Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Окуло-глоточная миотония. Это заболевание относительно распространено в Квебеке. Симптомы обычно появляются около 40 или 50 лет. Первые признаки болезни проявляются опустившимися веками, за которыми следуют слабость мышц глаз, лица и горла (глотки), вызывая трудности с глотанием пищи. Прогрессирование заболевания происходит медленно.
Исследования и прогресс
С 2005 года для лечения пациентов с развивающимся поражением мышц все чаще используются стволовые клетки. Для лечения мышечной дистрофии этим методом могут быть рассмотрены различные варианты заболевания, такие как: мышечные дистрофии Дюшенна, Беккера, миопатия поясничного и плечевого отдела.
Целью лечения является регенерация потерянных и поврежденных мышечных волокон с использованием регенеративного потенциала стволовых клеток. Для этого большое количество стволовых клеток вводится при помощи нескольких внутривенных и внутримышечных инъекций, что позволяет лучше нацеливать терапию именно на пораженную группу мышц.
Возможный прогресс
Терапия с применением стволовых клеток может обеспечить улучшение в плане мышечной массы, силы, движений, баланса, тремора и ригидности мышц. Стволовые клетки также могут замедлить будущую потерю мышечного объема и уменьшить симптомы.
Важно отметить, что лечение не является окончательным лекарством от этого заболевания и никоим образом не может решить проблему потери мышечных волокон. По этой причине прогресс после такого лечения не может быть постоянным. Исследования в этой области все еще ведутся.
Семейства заболевания
Обычно существуют два основных семейства МД:
- Мышечна врожденная дистрофия (ВМД), которая выражается в первые 6 месяцев жизни, сопровождает около десяти форм патологий переменной тяжести, включая ВМД с первичной недостаточностью мерозина, синдром Ульриха и Уокера-Варбурга;
- Мышечные дистрофии, появляющиеся в детстве или в зрелом возрасте:
![]()
Миопатия Дюшенна- Миопатия Беккера
- Миопатия Эмери-Дрейфуса (существует несколько форм)
- миопатия Ландузи-Дежерина
- Так называемая миопатия поясничного отдела — затрагивает мышцы вокруг плеч и бедер.
- Миотонические дистрофии (типы I и II), которые включают болезнь Штейнтера. Они характеризуются миотонией — когда мышцы не могут нормально расслабиться после сокращения.
- Окулофарингеальная миопатия
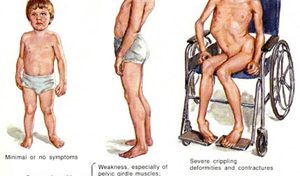
Эволюция дистрофии
Эволюция (развитие заболевания) МД сильно варьируется от одной формы к другой, а также от одного человека к другому. Некоторые формы быстро развиваются, что приводит к ранней утрате подвижности и ходьбе, а иногда и к смертельным сердечным или респираторным осложнениям, в то время как другие развиваются очень медленно — в течение десятилетий. Большинство врожденных мышечных дистрофий, например, которые мало выражены или почти незаметны, позже могут проявятся внезапно и с серьезными последствиями.
Возможные осложнения
Осложнения сильно различаются в зависимости от типа патологии. Некоторые нарушения могут затрагивать респираторные мышцы или сердце, иногда с очень тяжелыми последствиями.
Таким образом, сердечные осложнения довольно распространены, особенно у мальчиков с мышечной дистрофией Дюшенна.
Кроме того, дегенерация мышц заставляет тело и суставы деформироваться постепенно: на фоне этого у больных может развиваться сколиоз. Часто наблюдается сокращение мышц и сухожилий, что приводит к их стягиванию. Все эти нарушения приводят к деформации суставов: ноги и руки повернуты внутрь и вниз, деформируются колени или локти.
Также известно, что болезнь сопровождается тревожными или депрессивными расстройствами, поэтому больным требуется много внимания и поддержки, в первую очередь со стороны близких.
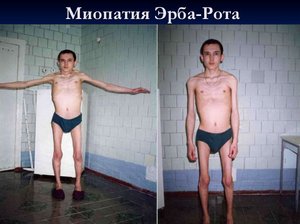
Миопатией называется заболевание преимущественно наследственного происхождения, в основе которого лежит дегенеративное поражение мышц с их прогрессирующей атрофией.
Это редкое заболевание, которое, тем не менее, встречается повсеместно, поражая людей вне зависимости от региона проживания и расовой принадлежности. По имеющимся данным, с псевдогипертрофической миопатией, или миопатией Дюшенна рождается каждый 3500-ый мальчик.
Причины и факторы риска
Первичная миопатия – генетически обусловленное заболевание. Непосредственный механизм, запускающий цепь патологических реакций и приводящий к нарушению метаболизма в мышечных клетках, пока невыяснен. Согласно существующей теории дефектных мембран, первичная патология кроется в плазматической мембране мышечных волокон, что приводит к бесконтрольному проникновению кальция, активирующего ферменты (протеазы), которые разрушают мышечную ткань. Происходит повышение уровня креатинфосфокиназы, что приводит к неспособности мышц удерживать и связывать креатин; уменьшается содержание АТФ (аденозинтрифосфат, главный донор клеточной энергии), результатом чего является атрофия мышечных волокон.
Нередко проявления миопатии возникают (а также усиливаются уже имеющиеся) под влиянием неблагоприятных провоцирующих факторов, к которым относятся:
- инфекционные заболевания;
- интоксикации;
- физическое и психоэмоциональное перенапряжение.
Приобретенные формы миопатии обусловлены заболеваниями, вызывающими повреждение нервов, обменными расстройствами, токсическим воздействием, интенсивным воспалительным или опухолевым процессом.
Формы заболевания
Существует несколько классификаций заболевания, но ни одна не считается полной.
Различают миопатии первичные и приобретенные. В большинстве случаев, когда речь идет об этой болезни, подразумевается наследственная, или первичная миопатия.
Приобретенная миопатия в зависимости от происхождения бывает:
- тиреоидная – может сопровождать как гипер-, так и гипотиреоз;
- стероидная – вызывается неадекватным приемом стероидсодержащих лекарственных препаратов (особенно фторсодержащих кортикостероидов) по поводу другого заболевания;
- алкогольная – определяется алкоголизм в анамнезе, заболевание часто сопровождается кардиомиопатией, в отличие от других форм может присутствовать болевой синдром;
- полимиозит (в свою очередь, может быть идиопатическим или вторичным).
Наследственные миопатии подразделяют на:
- мышечные дистрофии – им свойственно первичное поражение мышечных волокон;
- амиотрофии (спинальные и невральные) – вызваны нарушением иннервации мышц вследствие поражения центральной (спинномозговые моторные нейроны) или периферической нервной системы.
Наследственные миопатии с клинико-генетической точки зрения принято делить на три большие группы:
- псевдогипертрофическая, или миопатия Дюшенна.
- плече-лопаточно-лицевая, или миопатия Ландузи – Дежерина.
- ювенильная, или миопатия Эрба.
Позже к этим классическим формам добавились новые:
- лопаточно-перонеальная амиотрофия Давиденкова;
- дистальная поздняя наследственная миопатия Веландера;
- офтальмологическая;
- доброкачественная Х-хромосомная, или миопатия Беккера;
- врожденная (конгенитальная) непрогрессирующая;
- болезнь Мак-Ардла; и другие, более редкие формы.
Симптомы миопатии
Рецессивный тип наследования, сцепленный с Х-хромосомой, болеют только мальчики, заболевание передается от внешне здоровой матери.
Миопатия у ребенка дебютирует рано (первые 5 лет жизни), вначале в атрофический процесс вовлекаются мышцы таза и нижних конечностей с развитием выраженной псевдогипертрофии икроножных мышц. Примерно у трети пациентов наблюдается снижение интеллекта (легкая дебильность), не имеющая тенденции к прогрессированию. В сыворотке крови определяется высокий уровень альдолазы, трансаминазы и креатинкиназы. У матерей-носительниц патологического гена также повышены уровни сывороточных энзимов. Заболевание быстро и неотвратимо прогрессирует, приводя к летальному исходу от дистрофии или респираторных инфекций. Больные редко живут дольше 20 лет.
Аутосомно-доминантный тип наследования.
Может дебютировать в любом возрасте, но чаще начинается в подростковом. Характерна проксимальная атрофия на верхних конечностях и дистальная – на нижних. Один из специфических симптомов – крылоподобные лопатки. Может развиваться выраженный лордоз поясничного отдела позвоночника. Наблюдается Легкое или умеренное нарушение чувствительности в дистальных отделах конечностей.
Аутосомно-рецессивный тип наследования.
Наиболее частый тип наследственной миопатии. Атрофия начинается с мышц тазового пояса и нижних конечностей, имеет склонность к генерализации и рано приводит к обездвиженности больных.
Доминантный тип наследования.
Дебютирует после 20 лет. Атрофия начинается с мелких мышц кистей и стоп, затем затрагивает мышцы предплечий и голеней. Прогрессирует медленно.
Наследование может быть как аутосомно-доминантным, так и аутосомно-рецессивным, в последнем случае заболевание протекает тяжелее.
Часть случаев попадает под определение хронической прогрессирующей офтальмоплегии, в других к начальному двустороннему птозу век и глаз присоединяется атрофия мышц мягкого неба, гортани, языка, жевательных и мимических мышц.
Тип наследования рецессивный, сцепленный с Х-хромосомой. Сыновья людей, страдающих этой формой миопатии, свободны от патологического гена, а дочери – гетерозиготные носители.
Имеет сходство с офтальмологической формой, но начинается позже, прогрессирует медленнее, имеет доброкачественное течение.
Тип наследования в основном рецессивный.
Характеризуются врожденной мышечной гипотонией. Течение доброкачественное. Это группа миопатий, которые требуют дальнейшего изучения.
Разные типы наследования.
Дебют в любом возрасте, однако в подавляющем большинстве случаев – ранний (при рождении или в первые месяцы жизни), генерализованная мышечная гипотония, атрофия мышц, отсутствие или снижение сухожильных рефлексов, структурные скелетные аномалии.
Общими для большинства миопатий являются следующие симптомы:
- Слабость мышц появляется и медленно нарастает, имея двусторонний (симметричный) характер.
- Парестезии (нарушения чувствительности в виде покалываний, жжения, мурашек) в конечностях отсутствуют.
- Отсутствие болезненности в мышцах, несмотря на нарастающую слабость.
- В большей степени слабость затрагивает проксимальные (верхние) отделы конечностей (дистальные, или нижние чаще страдают при полинейропатии различной этиологии). Это проявляется тем, что становится затруднительным подъем по лестнице, расчесывание, умывание.
- Отсутствие нарушений функционирования тазовых органов.
- Отсутствие нарушений глубоких рефлексов (изредка могут быть незначительно понижены).
- Слабость лицевых мышц проявляется в невозможности вытянуть губы для свиста или поцелуя, невозможности зажмурить (или, напротив, широко открыть) глаза.
Диагностика
Возможно антенатальное выявление патологии по результатам исследования околоплодных вод (амниоцентеза).
Диагностика семейных форм и типичных случаев миопатии обычно не представляет сложности. Клиническое обследование выявляет мышечную слабость, как правило, изменяющуюся в течение дня. Пациента просят надуть щеки, вытянуть губы, зажмуриться, а затем быстро открыть глаза (вызывает затруднение). Проверяют наличие двоения в глазах, дизартрии, дисфагии. Исследуется сила подвздошно-поясничных и дельтовидных мышц, мышц сгибателей и разгибателей шеи (сгибатели более слабые, чем разгибатели).
Характерен так называемый миотонический феномен: больной, изо всех сил сжав кулак, не может его разжать.
В сомнительных случаях прибегают к следующим методам диагностических исследований:
- игольчатая электромиография – определяется миопатический паттерн, для которого характерна сниженная амплитуда М-ответа, при этом усиленная интерференция и полифазность потенциала;
- магниторезонансная томография (МРТ) мышц – определяется жировое и/или соединительнотканное перерождение мышечных волокон;
- биопсия мышц – позволяет подтвердить диагноз и дифференцировать ряд миопатий;
- исследование креатин-креатининового обмена – для миопатии характерно наличие креатинурии, повышение уровня креатинкиназы в сыворотке крови (особенно при псевдогипертрофической миопатии);
- генетическое исследование, включая молекулярно-генетический анализ, генетическое тестирование членов семьи, особенно способных к деторождению.
При приобретенных миопатиях диагностический поиск зависит от первопричины, например, при тиреоидной форме проводят определение уровня гормонов щитовидной железы.
Врожденные миопатии у детей следует дифференцировать с атонико-астатической формой детского церебрального паралича (ДЦП) и диспластическими кифосколиозами неясной этиологии.
Лечение миопатий
В настоящее время не существует возможности излечения врожденной миопатии, однако адекватная терапия помогает справиться с симптомами заболевания и замедлить его прогрессирование. Пациенты с миопатией должны наблюдаться терапевтом, неврологом, кардиологом и ортопедом. Лечение длительное, в большинстве случаев пожизненное. Пациенты получают его как амбулаторно, так и в стационаре. Периодически проводится контроль путем проведения обследования, и при необходимости в схему терапии вносятся изменения.
Лечение наследственных миопатий ставит своей задачей:
- Коррекцию обменных процессов и микроциркуляции в мышцах и нервной системе.
- Нормализацию функций нервной системы.
С этой целью могут быть назначены анаболические стероиды, трофотропные препараты, биогенные стимуляторы, витамины, препараты калия, антихолинэстеразные и вазоактивные средства.
Длительность каждого комплексного курса составляет 4–6 недель; он может включать, например, следующие препараты:
- Альфа-токоферол в виде внутримышечных инъекций по 0,3–1 мл 30 дней;
- АТФ (аденозинтрифосфорная кислота) внутримышечно по 1 мл 30 дней;
- Глутаминовая кислота – 0,5–1 г 3 раза в день длительно;
- Нивалин 0,25% раствор 0,1–2 мл в зависимости от возраста;
- Прозерин 0,05% раствор подкожно 1 мл 30 дней;
- Дибазол 1% 1 мл;
- Оксазил;
- Витамины группы В (В6, В12);
- АКТГ по 5-10 ЕД 4 раза в день в течение двух недель при ретракции мышц.
Хороший терапевтический эффект демонстрируют гемотрансфузии, переливают по 100-150 мл крови 1 раз в неделю на протяжении 4–6 недель.
Используются следующие методы:
- ионофорез с кальцием;
- электрофорез;
- рентгенотерапия диэнцефальной области;
- ультразвуковое воздействие на мышцы;
- электростимуляция мышц;
- массаж;
- пассивное растягивание сухожилий.
Применяется лечебная физкультура (ЛФК). Регулярные упражнения способны замедлить прогрессирование слабости и контрактуры, характерной для поздних стадий миопатии, однако требуется соблюдать осторожность при их выполнении, чтобы избежать перенапряжения ослабленных мышц. Эффективным вариантом адекватной физической нагрузки является плавание в бассейне под наблюдением инструктора по ЛФК (физиотерапевта).
В ряде случаев показана ортопедическая коррекция деформаций позвоночника с помощью специальных облегченных корсетов или ортопедической обуви.
В ряде случаев целесообразно хирургическое лечение, заключающееся в удлинении укоротившихся сухожилий.
Одним из важных элементов лечения является психологическая поддержка пациентов как семейная, так и профессиональная. Их близкие также должны получать психологическую помощь.
Постоянно ведется поиск новых методов и эффективных средств лечения как наследственных, так и приобретенных форм заболевания. Актуальные направления – клеточная и генная терапия. Миопатии, связанные с обменными нарушениями, довольно успешно лечатся при помощи средств, восполняющих недостаток того или иного фермента. Так, новый препарат Трансларна продемонстрировал высокую эффективность в лечении болезни Дюшенна.
Лечение приобретенных миопатий, или миопатических синдромов в целом такое же, однако основной задачей является устранение основной патологии. Например, важным условием лечения алкогольной миопатии является полный отказ от употребления алкоголя и дезинтоксикация, тиреоидной – коррекция тиреоидных гормонов, стероидной – отказ от причинных лекарственных препаратов.
Возможные осложнения и последствия
Последствия миопатии связаны с нарушением функций разных групп мышц, например, кардиомиопатия приводит к сердечной недостаточности, миопатия, затрагивающая межреберные мышцы – к ухудшению работы легких со всеми вытекающими последствиями. Развиваются неврологические расстройства. Постепенно организм слабеет, снижаются функции иммунной системы, из-за чего пациенты страдают частыми инфекционными заболеваниями, которые в некоторых случаях и являются непосредственной причиной смерти.
Прогноз
Прогноз зависит от формы заболевания. Он неблагоприятен в отношении псевдогипертрофической миопатии, которая быстро приводит к тяжелой инвалидности и ранней смерти. Прогноз для миопатий, дебютирующих позже, в конце подросткового периода, благоприятнее.
Профилактика
Предотвратить появление врожденной миопатии не представляется возможным. Выявление дефектного гена может помочь в планировании беременности людям, чей семейный анамнез связан с наследственными формами заболевания.
Профилактические меры эффективны для приобретенных миопатий. Они заключаются в отказе от самолечения, отказе от злоупотребления алкоголем, своевременном выявлении и лечении заболеваний щитовидной железы, профилактике интоксикаций.
Видео
Предлагаем к просмотру видеоролик по теме статьи.
Миопатией названа целая группа заболеваний мышечной ткани, преимущественно – скелетной мускулатуры, которая характеризуется стойким и прогрессирующим обменом веществ в ней. В результате этих нарушений мышцы постепенно теряют свою функцию, вплоть до полного поражения — у человека развивается вначале слабость мышц, вплоть до полного ограничения подвижности и невозможности вести активную жизнь.
Как проявляется миопатия на ранних и поздних стадиях заболевания?
Заболевание миопатия развивается постепенно, и на первых стадиях симптомы болезни могут быть стерты. Слабость в мышцах человек может списывать на усталость, последствия других заболеваний и т.д.
В самом начале развития миопатии врачи также могут неверно истолковать симптомы и ставить совершенно другие диагнозы.

- Слабость в мышцах, которая носит постоянный характер. После отдыха слабость остается, либо может уменьшаться, но – незначительно.
- Дальнейшее развитие миопатии характеризуется истончением, или атрофией, мышц – они становятся малоподвижными и тонкими. В результате заболевания могут страдать, как отдельные мышцы или группы мышц (например, только мышцы бедра или мышцы верхнегрудного отдела), так и все мышцы тела одновременно.
- Мышечный тонус сильно снижен – мышцы могут быть очень вялыми, появляется их дряблость.
- Вследствие слабости мышечного корсета в результате миопатии у человека возникает искривление позвоночного столба – это может быть кифоз, лордоз, сколиоз, которые со временем прогрессируют.
- На фоне истонченных мышц на отдельных участках тела, мышцы с нормальной функцией выглядят увеличенными – наблюдаются так называемые псевдогипертрофии.
- Сбор анамнеза.
- Неврологический осмотр и оценка мышечного тонуса.
- Оценка походки, рефлексов конечностей.
- Диагностика деформаций костей скелета, особенно – позвоночника.
- Лабораторные исследования – анализ крови на уровень креатинкиназы и гормонов щитовидной железы.
- Биопсия мышц – гистологическое исследование на факт дистрофии мышечного волокна.
- Генетические исследования с целью выявить наследственные факторы развития заболевания.
Причины возникновения миопатии
Среди основных факторов, которые являются причиной возникновения миопатий, известны следующие:
- Наследственная природа миопатии.
- Генетический дефект — вследствие недостаточности одного из важных ферментов, обеспечивающих нормальный обмен веществ в мышцах.
- Нарушения гормональной системы – например, тиреотоксикоз.
- Различные системные заболевания и патологии соединительной ткани– например, склеродермия.
По причинам возникновения заболевания различаются миопатии первичные и вторичные:
Первичные миопатии – это самостоятельно возникающие заболевания (чаще всего — наследственные).
К этой группе относятся:
- Врожденные миопатии (малыш рождается слабым, не способен громко кричать и хорошо брать грудь).
- Ранние детские миопатии (проявляются в возрасте 5-10 лет).
- Юношеские миопатии – возникают в пубертатном периоде.
Вторичные миопатии являются следствием и симптомом других заболеваний или состояний – например, интоксикации, нарушениях эндокринной системы).
По степени слабости мышц различают:
- Преимущественно проксимальные миопатии– проявляются слабостью мышц, которые на конечностях находятся ближе к туловищу (бедро, плечи).
- Дистальные миопатии – слабость наблюдается мышцах, расположенных дальше от тела (икроножные, предплечья и кисти рук).
- Смешанные миопатии, которые сочетают в себе дистальные и проксимальные миопатии.
- Дыхательная недостаточность, когда заболевание затрагивает дыхательную мускулатуру.
- Ограничение или полная потеря способности двигаться.
- Застойная пневмония, что является следствием малоподвижности и поражения дыхательной мускулатуры.
- Искривления позвоночника, межпозвонковые грыжи.
- Парезы, параличи.
Современные методы лечения
Наследственная миопатия не поддается излечению – возможно только ослабить основные симптомы и прогрессирование заболевания. Как правило, для поддержания позвоночника больному назначают ортезы. При миопатии показаны специальная лечебная гимнастика, дыхательная гимнастика.
При миопатии эндокринной природы — при гиперфункции щитовидной железы — больному назначается лечение антагонистами тиреоидных гормонов – тиреостатиками.
Вторичная миопатия вследствие заболеваний соединительной ткани (склеродермия) нуждается в лечении цитостатиков и стероидных гормонов.
При всех видах миопатии больным назначается белково-минеральная диета, из процедур наиболее эффективными являются массаж, растирания пораженных участков, бальнеолечение, ванны.
Методов оперативного лечения миопатии нет, потому что заболевание прогрессирует в любом случае. Единственно правильный путь – назначение больному ортопедических корсетов в самом начале заболевания для поддержания позвоночника и профилактики его искривления. Корсеты могут индивидуально разрабатываться и подбираться врачом-ортопедом.
Читайте также:
