
Генная терапия при мышечной дистрофии
ДЛЯ ТОГО ЧТОБЫ СКАЧАТЬ СТАТЬЮ В ФОРМАТЕ PDF ВАМ НЕОБХОДИМО АВТОРИЗОВАТЬСЯ, ЛИБО ЗАРЕГИСТРИРОВАТЬСЯ
В обзоре обсуждаются современные подходы к лечению мышечных дистрофий - гетерогенной группы нервномышечных заболеваний, которые проявляются в виде прогрессирующей слабости мышц, а также их частичной потери, что во многих случаях приводит к смерти. Эффективных медикаментозных способов лечения дистрофий в настоящее время нет. Однако существует несколько исследуемых терапевтических вариантов лечения мышечных дистрофий, включающих генную терапию и трансплантацию миогенных клеток-предшественников - клеточную терапию. В статье обсуждаются достижения и трудности на пути внедрения генной терапии в клиническую практику лечения мышечных дистрофий. Особое внимание уделено клеточной терапии, перспективному направлению регенеративной медицины, дающему надежду на излечение многих ранее не излечимых как наследственных, так и приобретенных заболеваний. Обсуждаются потенциальные источники соматических и эмбриональных стволовых/прогениторных клеток человека, которые могут использоваться как в качестве объектов приложения генно-инженерных методов, так и для клеточной терапии мышечных дистрофий. Обсуждаются проблемы, стоящие на пути успешного внедрения в клиническую практику лечения мышечных дистрофий стволовых/проге-ниторных клеток человека.
Введение
Мышечная дистрофия - это группа наследственных заболеваний, которые характеризуются прогрессирующей мышечной слабостью и потерей мышечных тканей. В настоящее время все дистрофии считаются заболеваниями неизлечимыми. В некоторых случаях болезнь приводит к смерти. Мышечные дистрофии различаются по группам поврежденных мышц, возрасту, при котором начинается болезнь, и скорости ее прогрессирования. Молекулярной причиной, лежащей в основе многих дистрофий, являются мутации в генах, кодирующих компоненты дистрофин-гликопротеинового комплекса, который связывает миофибрильный цитоскелет с внеклеточным матриксом 3.
Наиболее часто встречается и наиболее тяжело протекает мышечная дистрофия Дюшенна (МДД). МДД поражает мальчиков с частотой, равной 1 на 3500. Она проявляется в 1-2-летнем возрасте и характеризуется потерей независимого передвижения уже к 10 годам. Больные МДД, как правило, умирают в возрасте 15-25 лет от сердечной или легочной недостаточности вследствие атрофии дыхательных и сердечной мышц. Сходными симптомами и причиной возникновения характеризуется мышечная дистрофия Беккера (МДБ), однако протекает это заболевание в менее тяжелой форме. Обе болезни являются следствием мутаций в гене дистрофина, который локализован на коротком плече Х-хромосомы и является самым большим из известных генов человека. Он содержит 79 экзонов и составляет приблизительно 0,1 % всего генома человека 4. Этот ген кодирует мышечный белок дистрофин с молекулярным весом 427 кДа, который является основным структурным белком, стабилизирующим сарколемму мышечного волокна. Своим N-концом он связывается с актином - главным компонентом внутриклеточного цитоскелета, а С-концом - с группой дистрофин ассоциированных дистро- и саркогликанов -белков сарколеммы, и через них - с основным белком внеклеточного матрикса - ламинином (рис. 1).
Мутации в гене дистрофина приводят к полному отсутствию или сильному снижению уровня дистрофина в случае МДД, либо к образованию частично функционального дист-рофина в случае МДБ. Отсутствие или функциональная недостаточность этого белка приводит к нарушению целостности клеточной мембраны и является причиной запуска процессов дегенерации мышечного волокна. Результатом этого процесса является повышение проницаемости мембран и возникновение в них физических разрывов, что приводит к выходу ферментов из мышц в сыворотку крови (повышение в сыворотке крови активности креатинкиназы является биохимическим маркером МДД и МДБ). На начальных стадиях заболевания дегенерация мышечных волокон компенсируется активной регенерацией фибрилл, благодаря делению и слиянию мышечных сателлитных клеток. Однако с возрастом этот процесс становится менее эффективным, что приводит к прогрессирующей мышечной слабости [7].
В настоящее время при отсутствии эффективных медикаментозных способов возлагаются большие надежды на использование для лечения мышечных дистрофий методов генной и клеточной терапий. Задачей этих способов лечения является восстановление синтеза дистрофина в мышечных клетках, что достигается либо путем встраивания в клетки нормального гена дистрофина, либо коррекцией мутаций в самом гене или в иРНК [8].
Генная терапия мышечных дистрофий
Основное различие генной и клеточной терапий заключается в способе доставки нормального гена дистрофина в мышечную клетку. Генно-инженерные подходы доставки гена используют, как правило, вирусные векторы. В экспериментах на мышах была продемонстрирована достаточно эффективная и долговременная трансфекция скелетных и сердечной мышц, а также мышц диафрагмы после введения аденовирусных, ретровирусных или лентивирусных векторов, доставляющих функциональный ген дистрофина или минидистрофина 10.
Вместе с тем, использование вирусных носителей, особенно в экспериментах in vivo, наталкивается на существенные методические трудности. К ним, например, относятся недостаточная пакующая способность ретровирусов и необходимость наличия клеток-хелперов. Наибольшим препятствием использования вирусных векторов для доставки генетических конструкций является выраженный иммунный ответ реципиента на вирусные антигены. Несмотря на огромный объем работ по модификации генома вируса-носителя и сокращению его размера до минимально возможного, иммунный ответ, тем не менее, сохраняется и делает бессмысленным повторное введение генно-инженерных конструкций. В результате оказывается невозможно достичь такого уровня трансфекции, при котором наступает эффект лечения. Большинство исследователей полагает, что достижение терапевтического эффекта возможно при успешной трансфекции не менее 20% [по последним данным - даже 40%) всех мышечных волокон, включая мышцы сердца и диафрагмы. При этом основными критериями эффективности трансфекции являются появление дистрофин-положительных мышечных волокон, нормализация уровня креатинкиназы в сыворотке крови, изменение физиологических параметров [силы мышц и др.).
Существуют также невирусные способы доставки к ДНК гена дистрофина, которые включают в себя баллистическую трансфекцию, методы электропорации [электрошока), введение генетических конструкций в составе липосом, либо упакованных с помощью олигопептидов, молекулярных конъюгатов или полимерных носителей [9, 13, 14]. Эти способы не вызывают такого иммунного ответа, как вирусные векторы, однако способность к трансформации у большинства из них ниже.
Несмотря на довольно интенсивные исследования на животных моделях, до настоящего времени клинические испытания генно-инженерных методов лечения дистрофий на больных людях практически не проводились.
Следует также упомянуть о существовании еще одного подхода к лечению дистрофий - активации синтеза в мышечных клетках репрессированного в процессе онтогенеза аутосомного гомолога гена дистрофина - гена утрофина [15]. Известно, что в эмбриогенезе человека приблизительно до семи недель развития дистрофин не экспрессируется, и его функцию в мышцах выполняет белок утрофин [16]. Между 7 и 19 неделями развития экспрессируются оба белка, после чего происходит замещение мышечного утрофина на дистрофин. Эти белки похожи своими N- и С-концевыми доменами, играющими решающую роль в функции дистрофи-на, тогда как функционально малозначимый центральный rod domain присутствует в утрофине в сильно укороченном виде. Если достичь дерепрессии гена утрофина у больных мышечной дистрофией, то продукт его экспрессии белок утрофин мог бы компенсировать недостаток дистрофина в мышцах [17]. В настоящее время получены данные, свидетельствующие о том, что in vivo трансфекция mdx мышей геном утрофина приводит к экспрессии этого белка в скелетных мышцах и диафрагме 18. Эти результаты указывают на принципиальную возможность коррекции недостатка дистрофина в мышечных волокнах с помощью утрофина.
Клеточная терапия мышечных дистрофий
Перспективным направлением клеточной терапии мышечных дистрофий представляется трансплантация как стволовых клеток [СК), способных к самовозобновлению и дифференциации во множество клеточных линий организма, так и миогенных прогениторных клеток. При этом рассматриваются возможности использования для трансплантации как взрослых, так и эмбриональных стволовых/прогенитор-ных клеток.
Стволовые клетки костного мозга. В КМ, основываясь на адгезивных свойствах in vitro, различают два типа СК: адгезивные стромальные мезенхимальные СК и неадгезивные гемопоэтические СК. Мезенхимальные СК могут дифференцироваться в клетки кости, хряща, жировой ткани и мышц как in vitro, так и in vivo. Миогенные клетки могут быть получены in vitro при действии на адгезивные мезенхимальные клетки 5-азацитидина (5-azacytidine), который активирует мышечный переключатель MyoD [35]. Субпопуляция мезенхимальных СК также была идентифицирована в мо-нонуклеарных клетках КМ - это CD45+/glycophorin+ клетки [36]. После ex vivo экспансии эти мультипотентные взрослые прогениторные клетки могут быть трансплантированы в необлученный организм мышей, где они приживаются и развиваются в гемопоэтические линии, эпителиальные клетки, клетки печени, легких, кишечного эпителия. При введении в раннюю бластоцисту эти клетки дифференцировались в мышечные. И хотя их способность становиться предшественниками скелетной мышцы при внутривенном введении продемонстрирована не была, свойство этих мультипотент-ных взрослых прогениторных клеток активно пролиферировать без очевидного старения и потери мультипотенции делает их привлекательным кандидатом для клеточной терапии.
Более перспективными для клеточной терапии мышечных дистрофий являются неадгезивные гемопоэтические СК, которые, помимо гемопоэза, участвуют в формировании нервной ткани [37, 38], печени [39], сердечной мышцы [29] и скелетных мышц. Было показано, что эти миогенные прогениторы могут мигрировать и участвовать в регенерации поврежденной мышечной ткани после трансплантации КМ [40]. В этом исследовании также было продемонстрировано, что в репарации поврежденных кардиотоксином мышечных клеток могут участвовать не только клетки КМ, введенные непосредственно в мышцу, но также и введенные внутривенно. В обоих случаях донорские клетки сливались с мышечными волокнами реципиента. Эксперименты по внутривенному введению донорских клеток КМ mdx мышам также продемонстрировали, что трансплантированные клетки попадают из кровеносной системы в мышечную ткань и сливаются с волокнами дистрофических мышц mdx мышей [30, 41, 42]. Однако, во всех этих экспериментах репарация дистрофических мышц трансплантированными клетками КМ никогда не превышала 1 %. Это можно объяснить как слабым захватом клеток КМ, так и неспособностью стволовых клеток КМ адекватно реагировать на клеточные сигналы реципиента.
Несмотря на то, что использование клеток КМ для лечения модели МДД у животных оказалось неэффективным, существует мнение некоторых исследователей, что трансплантация КМ для лечения мышечной дистрофии у людей будет более эффективной. Способность СК КМ человека участвовать в репарации мышечных волокон была продемонстрирована Gussoni с соавторами [43]. Они представили иммуногистохимический и FISH анализ мышц мальчика, которому в возрасте 1 года для лечения тяжелого X-связанного иммунодефицита, диагностированного вместе с МДД, была проведена трансплантация КМ. При этом, в 12 лет МДД имела умеренное протекание, а в мышцах больного через 13 лет после трансплантации было обнаружено присутствие диплоидных донорских ядер [0,5-0,9% мышечных волокон), что указывает на способность экзогенных клеток КМ человека сливаться со скелетными мышечными волокнами реципиента и сохраняться на протяжении, по крайней мере, 13 лет.
Таким образом, несмотря на возможность взрослых ге-мопоэтических СК сливаться с дистрофическими скелетными мышечными волокнами, частота таких случаев слишком низка для того, чтобы быть эффективным способом лечения мышечных дистрофий.
Клетки пуповинной крови. Очень близки по свойствам клеткам КМ клетки пуповинной крови. В их составе также присутствуют как кроветворные, так и мезенхимальные СК [44]. Применение этих клеток лишено морально-этических ограничений, их легко получать, количество СК в пуповинной крови значительно больше, пролиферативный потенциал выше, они не отягощены грузом генетических мутаций, накопленных взрослыми соматическими СК КМ. Клетки пуповинной крови менее иммунологически зрелые. Эксперименты, проведенные на мышах [45], продемонстрировали, что миогенные прогениторы [предшественники) присутствуют в пуповинной крови человека и способны мигрировать и дифференцироваться в миоциты. Однако, как и в случае СК КМ, до сих пор отсутствуют доказательства эффективного терапевтического действия клеток пуповинной крови при их использовании для лечения мышечных дистрофий. Как и для всех остальных потенциальных источников клеток для лечения мышечных дистрофий, существуют методические проблемы, связанные с выделением из пуповинной крови популяции миогенных предшественников и их дальнейшей экспансией в условиях in vitro.
СатК были одним из первых типов клеток, используемых в клеточной терапии мышечных дистрофий. СатК, которые выращиваются in vitro, представляют собой мононуклеар-ные клетки, коммитированные по миогенному пути [миоб-ласты). Эксперименты по внутримышечной трансплантации миобластов, изолированных из мышей дикого типа, mdx мышам продемонстрировали экспрессию дистрофина in vivo [55, 56]. Эти исследования послужили основанием проведения клинических испытаний миобластов с целью лечения МДД 60. Для этого донорские СатК были изолированы в результате биопсии мышц у близких родственников больных детей. Затем эти клетки выращивались в условиях in vitro, после чего 80-100 миллионов донорских миобластов было введено в мышцы детей путем множественных инъекций. Контрлатеральные мышцы были инъецированы как контроль плацебо. Однако, иммуногистохимические и RT-PCR исследования мышц больных, которые проводились на протяжении года, показали, что трансплантация миобластов таким способом была неэффективна. Несмотря на то, что пересаженные миобласты действительно сохранялись и продуцировали дистрофин в мышечных волокнах пациентов с МДД, частота, при которой это встречалось, была очень низкой (
16 февраля 2016
- 3800
- 3,1
- 0
- 6
Принцип генной терапии миодистрофии Дюшенна/Беккера. Миодистрофию Дюшенна (МДД) вызывают мутации гена дистрофина (DMD), приводящие к сдвигу рамки считывания, а более мягкую миодистрофию Беккера (МДБ) — мутации без смещения рамки считывания. Лечения этой болезни пока нет. Генная терапия поможет улучшить или даже восстановить функции мышц.
![]()
Анна Петренко

![]()
Антон Чугунов![]()
Ольга Волкова


- CRISPR/CAS
- Генная инженерия
- Медицина
Мышечная дистрофия Дюшенна — тяжелейшее Х-связанное заболевание, эффективного лечения которого до сих пор нет. В одном из последних номеров Science вышли целых три статьи об успешном тестировании на мышиных моделях технологии CRISPR/Cas9 для лечения этой болезни. Может быть, у этого подхода есть шанс добраться и до клиник?
Мышечная дистрофия Дюшенна, от которой страдает один из 3600-5000 новорожденных мальчиков, вызывается отсутствием дистрофина — белка, который соединяет цитоскелет и внеклеточный матрикс в мышечном волокне и обеспечивает его стабильность при сокращении (рис. 1). Из-за мутаций гена DMD рамка считывания при трансляции его мРНК сдвигается, и синтез белка преждевременно прекращается. Врожденная болезнь очень быстро прогрессирует: ее диагностируют в возрасте около четырех лет, а к 10 годам ребенку обычно уже нужна инвалидная коляска. Это происходит потому, что без дистрофина волокна повреждаются, и как только регенеративная способность мышечных волокон исчерпывается, они заменяются фиброзной и жировой тканями [1]. Как показывают исследования, когнитивные функции у ребенка тоже могут быть нарушены [2]. Больше 30 лет с таким заболеванием, как правило, не живут, а смерть наступает от сердечных и респираторных осложнений. Более мягкий вид миодистрофии, связанной с геном DMD, — это мышечная дистрофия Беккера, когда мутации не приводят к смещению рамки считывания [3].
Дистрофин находится на внутриклеточной поверхности сарколеммы вдоль всей длины мышечных волокон и входит в состав дистрофин-ассоциированного гликопротеинового комплекса (ДАГК, DGC). Он связывается одним концом с F-актином цитоскелета, а другим — с β-дистрогликаном, что стабилизирует волокна во время сокращения. Ген дистрофина — один из самых длинных у человека.
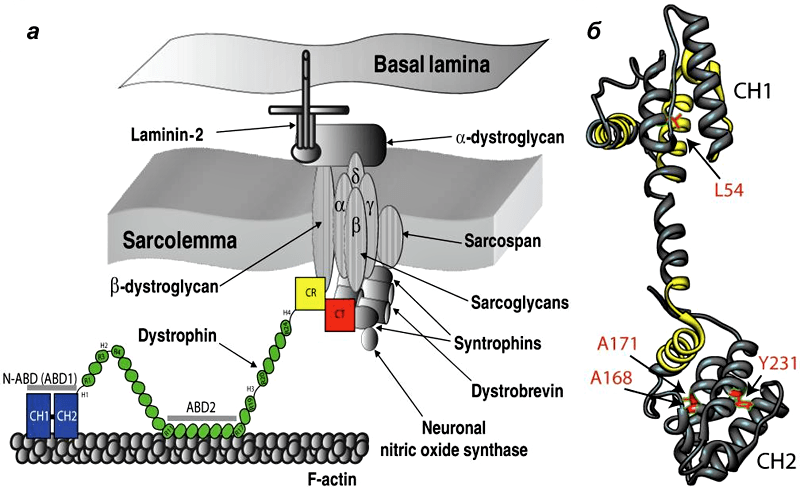
Рисунок 1. Мутации в дистрофине — причина развития миодистрофии Дюшенна. а - Дистрофин связывается с актиновыми филаментами (часть цитоскелета) через домены N-ABD и ABD2) и с ДАГК через домены CR и CT. б — Кристаллическая структура N-ABD дистрофина. Зоны связывания с актином показаны желтым, четыре хорошо изученных мутации, вызывающих заболевание, — красным.
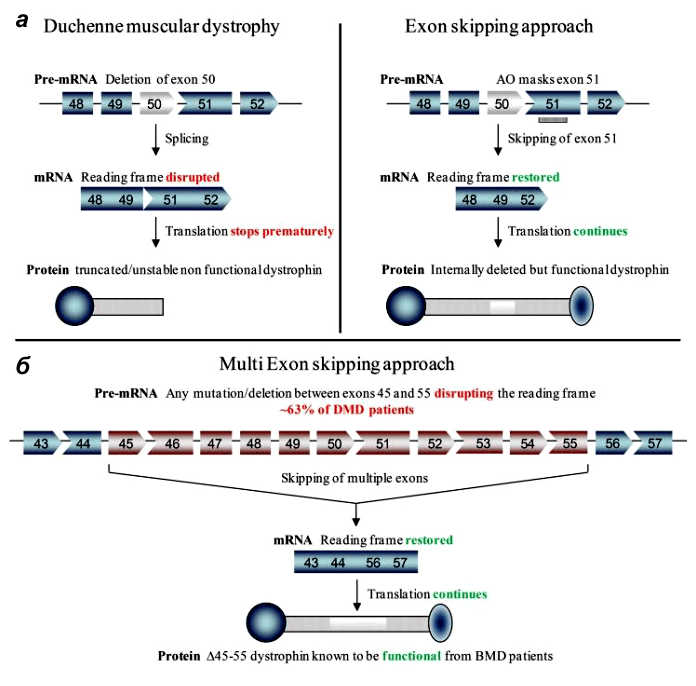
У стратегии удаления экзонов есть даже преимущества перед воссозданием полной длины гена: ее проще разработать, чем восстановить индивидуальные делеции каждого пациента [7].
Конкурирующие лаборатории: кто первым воплотит технологию в терапию для человека?
Ученые трех лабораторий успешно применили технологию пропуска экзонов in vivo на стандартном объекте — мышах — и показали, что их метод помогает восстановить рамку считывания и частично восстановить синтез дистрофина. Поскольку даже невысокий его уровень (3–15% от нормального) приносит терапевтическую пользу, результаты работ можно назвать успешными.
Группа Эрика Олсона уже не в первый раз использует метод CRISPR/Cas9 в своих работах по мышечной дистрофии Дюшенна. В 2014 году ученые исправили мутацию в зародышевой линии мышей и предотвратили развитие болезни. Однако, поскольку пренатальное редактирование генома на человеческих эмбрионах (пока?) запрещено, исследователям пришлось придумать способ постнатального применения технологии.
Группа Эми Уаджерс провела во многом похожий эксперимент [8]. После множества подготовительных этапов работы по редактированию генома и пропуску экзона на клетках и животных их опыт тоже увенчался успехом: программируемые CRISPR-комплексы в составе аденоассоциированного вируса (AAV) были доставлены с помощью локального и системного введения к дифференцированным скелетным волокнам, кардиомиоцитам и сателлитным мышечным клеткам новорожденных и взрослых мышей. Если редактирование направлено только на мышечные волокна, то эффект со временем может сойти на нет. Однако, как отмечает Уаджерс, редактирование генов в сателлитных клетках может обеспечить гораздо более длительный результат. Оно способно привести к созданию пула регенеративных клеток, несущих отредактированный ген дистрофина, и в результате обычной репарации мышц отредактированный ген окажется и в мышечных волокнах.
Терапия миодистрофии Дюшенна: старые и новые подходы
По словам Олсона, главное отличие новой стратегии с использованием вектора, вмещающего в себя компоненты для редактирования генома, от других терапевтических методов в том, что она устраняет причину болезни. А какие еще подходы разрабатывают ученые?
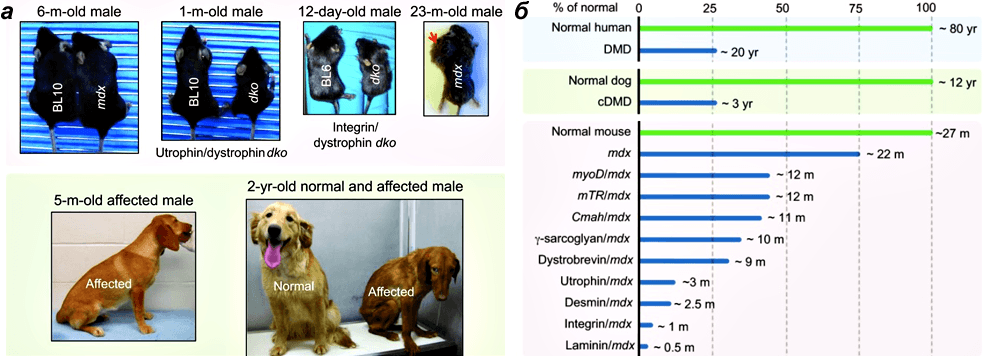
Рисунок 3. Животные модели миодистрофии Дюшенна. а — Проявления миодистрофии Дюшенна у мышей и собак. Вверху: у мышей mdx симптомы проявляются только в старости, и они склонны к образованию рабдомиосарком — опухолей мышечного происхождения. Размер мышей с нокаутами генов атрофина/дистрофина и интегрина/дистрофина значительно меньше, чем их ровесников дикого типа (BL10 и BL6). Внизу: проявления болезни у пятимесячной больной собаки. Различия между здоровой и больной двухлетними собаками. б — Сравнение продолжительности жизни здоровых и больных людей, собак и различных линий мышей.
Один из многообещающих подходов — это клеточная терапия. Хотя опыты с внутримышечной инъекцией миобластов от здоровых доноров провалились, технологии с использованием стволовых клеток и индуцированных плюрипотентных стволовых клеток (ИПСК) пока успешно испытываются на моделях не только миодистрофии Дюшенна, но и болезни Альцгеймера, Паркинсона, Хантингтона, спинальной мышечной атрофии, бокового амиотрофического склероза, аутизма и шизофрении [14–16]. Например, в 2013 году исследователи из Бостонской детской больницы (Boston Children’s Hospital’s Stem Cell Program) с помощью смеси трех малых молекул (форсколина, основного фактора роста фибробластов bFGF и ингибитора гликогенсинтазы киназы-3) перепрограммировали ИПСК из кожи пациентов с миодистрофией Дюшенна в мышечные клетки, которые затем успешно прижились у мышей. Сейчас из ИПСК получены кардиомиобласты и нейроны [2].
Другие исследования показывают, что восстановление нормального уровня синтеза оксида азота (NO), который снижается у больных из-за нарушения активности NO-синтазы (nNOS), ослабляет воспаление, повышает активность собственных стволовых клеток и реконструирует морфологию и функции скелетных мышц [3].
Уже в фазе II клинических испытаний находится препарат Givinostat — ингибитор гистондеацетилаз, который замедляет прогрессирование болезни в мышиной модели.
Такой массированный экспериментальный удар по миодистрофии Дюшенна вселяет надежду. Станет ли технология CRISPR/Cas9 ведущей в разработке терапии, которую смогут принять на вооружение клиницисты? Возможно, не за горами и публикации похожих работ по другим заболеваниям, где нужно избавиться от мутаций в одном-единственном гене? Это мы узнаем из ближайших выпусков Science (а также других почетных журналов).



Профессор Дуншэнь Дуань и его группа смогли смягчить симптомы мышечной дистрофии у собак с моделью мышечной дистрофии Дюшенна, используя методы генной терапии.
Результаты экспериментов по использованию методов генной терапии для лечения собак с моделью мышечной дистрофии Дюшенна (МДД), полученные учеными Университета Миссури, можно назвать технологическим прорывом и гигантским скачком вперед в лечении этого заболевания.
Причиной мышечной дистрофии является замещение пораженной мышечной ткани тканями других типов – фиброзной, костной или жировой. Мышечная дистрофия Дюшенна (МДД) – наиболее распространенный тип мышечной дистрофии, преимущественно поражающий мальчиков, – обусловлена генной мутацией, нарушающей биосинтез белка дистрофина, необходимого для выживания и функционирования клеток мышц. Отсутствие дистрофина запускает цепную реакцию, которая в конечном итоге приводит к дегенерации и гибели мышечных клеток. Найти ключ к восстановлению синтеза дистрофина ученые пытаются в течение многих лет, но им приходится сталкиваться с множеством проблем.
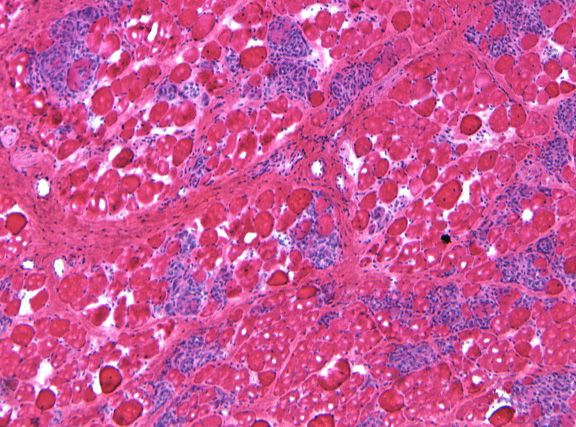
При отсутствии лечения у собак с моделью мышечной дистрофии Дюшенна наблюдается гибель мышечных клеток (округлые структуры) и выраженное воспаление (синее окрашивание).
Для преодоления этого препятствия группа во главе с профессором медицинских исследований Школы медицины Университета Миссури Дуншэнь Дуанем сконструировала новый ген микродистрофина, несущий важную функциональную область, которой не было в более раннем варианте этого гена.
Ученые поместили новый микроген в вирус, ввели вирус в мышцы собак с дистрофией, а затем оценили силу мышц и признаки их поражения, сравнив эти показатели с показателями животных контрольной группы. Тщательный анализ состояния 22 собак показал, что новая версия микродистрофина не только уменьшает воспаление и фиброз, но и значительно увеличивает мышечную силу.
Тем временем ученые Института сердца Лиллехея Университета штата Миннесота создали стволовые клетки, способные восстанавливать мышечную ткань в мышиной модели мышечной дистрофии Дюшенна. Достичь этого успеха им помогло сочетание двух передовых биотехнологических методов – генной инженерии и перепрограммирования клеточного генома.
Работа американских исследователей, доказавшая состоятельность самого принципа – сочетания технологии индуцированных плюрипотентных стволовых клеток с генетической коррекцией, – представляет собой значительный шаг в развитии методов лечения мышечной дистрофии Дюшенна и близких к ней заболеваний, основанных на аутологичных клетках. Ученые надеются, что она проложит путь к тестированию данного подхода на человеческих индуцированных плюрипотентных стволовых клетках, полученных из клеток пациентов с этими тяжелыми недугами.
Результаты исследования опубликованы в журнале Nature Communications.
Для достижения столь значимого результата в лечении мышечной дистрофии у мышей, исследователи использовали три новаторские технологии.
Во-первых, они перепрограммировали в плюрипотентные клетки, способные дифференцироваться в любые зрелые типы клеток организма, фибробласты животных. Перепрограммированные фибробласты несли мутации в генах дистрофина и атрофина, вызывающие у мышей развитие тяжелой формы мышечной дистрофии, очень близкой к человеческому заболеванию – мышечной дистрофии Дюшенна. Полученная таким образом модель хорошо имитировала то, что теоретически может происходить в организме человека.

Рита Перлингейро, PhD, адъюнкт-профессор Медицинской школы Университета штата Миннесота.
Третья использованная технология – метод получения стволовых клеток скелетных мышц из плюрипотентных клеток – процесс, разработанный лабораторией руководителя исследования Риты Перлингейро (Rita Perlingeiro), PhD.
Сочетание этих технологических платформ позволило ученым получить образующие мышцы стволовые клетки, которые не отторгаются иммунной системой организма. Образуя функциональные мышцы и реагируя на повреждение мышечных волокон, пересаженные клетки хорошо зарекомендовали себя у мышей с мышечной дистрофией.
К счастью, эти опасения ученых не подтвердились: эксперименты на мышах с поврежденными новыми мышечными волокнами показали, что клеточные трансплантаты обеспечили животных-реципиентов полностью функциональными мышечными стволовыми клетками.
Этот проект доказывает состоятельность концепции объединения технологии индуцированных плюрипотентных стволовых клеток и коррекции генома методами генной инженерии при лечении мышечной дистрофии.
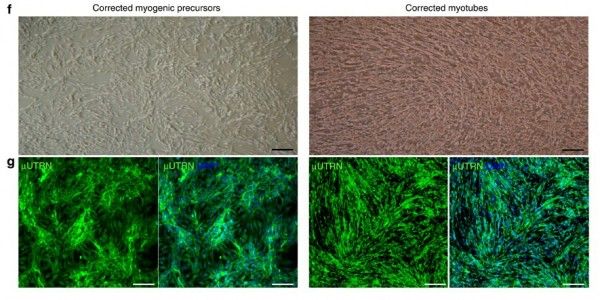
(f) Фазово-контрастные изображения монослоя культуры в условиях пролиферации (слева) и дифференциации (справа). (g) Иммунофлуоресцентное окрашивание μUTRN (зеленый) в пролиферирующих миогенных прогениторах, полученных из индуцированных плюрипотентных стволовых клеток, (слева) и в их производных – мышечных трубочках (справа). Клетки окрашены DAPI (синий).
Эксперименты американских ученых прокладывают путь к тестированию данного подхода на перепрограммированных человеческих плюрипотентных стволовых клетках, полученных из клеток пациентов с мышечной дистрофией.
Предметы
Последние несколько лет были захватывающим временем для всех тех, кто посвятил себя разработке методов лечения мышечной дистрофии с появлением ряда клинически применимых методов лечения. Недавняя работа Millay et al. 1 вносит важный вклад, поскольку он открывает новую область модификации заболевания путем уменьшения митохондриально-зависимого некроза, чтобы смягчить последствия мышечной дистрофии у мышей. Потенциально это также может быть полезно для других состояний, связанных с митохондриально-зависимым некрозом.
Мышечные дистрофии представляют собой группу наследственных нарушений, характеризующихся прогрессирующим истощением мышц, часто проявляющимся в раннем детстве. Наиболее распространенной является мышечная дистрофия Дюшенна (МДД), которая поражает 1 из 3500 мужских рождений. МДД вызван дефектами самого большого гена в геноме человека, который охватывает 2, 5 Мб. Этот ген кодирует большой белок цитоскелета, называемый дистрофином, который связывает цитоскелет и, следовательно, сократительный аппарат мышечной клетки с внеклеточным матриксом. Дистрофин присоединяется к цитоскелету посредством его амино-конца и через область, близкую к карбокси-концу, к β-дистрогликану, встроенному в клеточную мембрану. Это, в свою очередь, связано с α-дистрогликаном и, следовательно, с белками во внеклеточном матриксе, включая ламинин α2. Комплекс мембраносвязанных белков (саркогликанов) связан со стабилизированным β-дистрогликаном. Дефицит любого из ассоциированных или связывающих белков вызывает мышечную дистрофию (обзор в 2 ).
Современные методы лечения МДД являются симптоматическими и значительно улучшают продолжительность жизни и качество жизни, но мало способствуют предотвращению потери мышечной функции. Экспериментальные методы лечения, которые в настоящее время исследуются, делятся на три категории. Первый восстанавливает экспрессию дистрофина, чтобы остановить прогрессирование заболевания, а ряд подходов модифицирует процессинг гена или вводит рекомбинантную версию дистрофина. К ним относятся антисмысловой опосредованный пропуск экзонов, считывание преждевременных стоп-мутаций и вирусный перенос генов, которые в настоящее время находятся на стадии клинических испытаний. 3, 4 Второе включает избыточную экспрессию компенсирующих генов либо путем переноса генов, либо путем усиления экспрессии с использованием небольших молекул. Генетические кандидаты включают аутрофин, аутосомный гомолог дистрофина, интегрин α7 и IGF-1. 2 Последняя категория включает изменение процесса заболевания ниже по течению от дефицита дистрофина, и был протестирован ряд модифицирующих заболевание агентов, таких как блокада рецепторов ангиотензина II типа 1 (лозартан), блокада TNF-α и коэнзим Q10, и было показано, что эффективен в мышиной модели DMD. 5, 6 Результаты, представленные Millay et al. 1 являются важным дополнением к этой последней категории и представляют новую цель лечения.
При многих мышечных дистрофиях недостаток белка приводит к дестабилизации сарколеммы с соответствующим увеличением притока ионов кальция в мышечное волокно. В попытке удалить избыток кальция митохондрии становятся перегруженными, что вызывает увеличение проницаемости митохондрий. Если его не остановить, это приводит к некротической и / или апоптотической гибели клеток. Важным регулятором этого процесса является циклофилин D, и мыши, у которых отсутствует ген этого белка ( Ppif ), устойчивы к отеку, вызванному кальцием, и гибели клеток, вызванной ишемией / реперфузией. 7 Millay et al. демонстрируют, что скрещивание этих мышей Ppif - / - с мышами, у которых отсутствует δ саркогликан ( Scgd - / - , мышечная модель мышечной дистрофии конечностей 2F) или ламинин α2 ( Lama2 - / - , мышиная модель врожденной мышечной дистрофии 1A), заметно ослабил процесс заболевания, хотя и не предотвратил его полностью. Они также показали, что два раза в день в течение 6 недель лечение Debio-025, ингибитором циклофилина, значительно уменьшало заболевание у мышей MDX и Scgd - / - .
Debio-025 является синтетическим аналогом циклоспорина, не обладающим иммунодепрессивной способностью, но обладающим высокой ингибирующей способностью в отношении цис-транс- пролилизомеразной активности, ассоциированной с циклофилином А. Он разрабатывается Группой Debiopharm для лечения гепатита С и недавно завершил клиническое исследование Фазы IIa (//www.debiopharm.com/products/pipeline/debio-025.html). Результаты работы Миллея показывают, что использование Debio-025 предлагает новые стратегии лечения мышечных дистрофий, включая МДД; Однако важно быть осторожным при экстраполяции непосредственно от мыши к человеку. Ключевой проблемой было бы определение безопасной и эффективной дозы Debio-025 для человека. В большинстве доклинических исследований, в основном, доза для эффекта, в то время как дозы для человека обязательно обусловлены вопросами безопасности, а не максимальной эффективностью. Кроме того, любое успешное лечение мышечных дистрофий, вероятно, потребует непрерывных или повторных циклов лечения, и мало что известно о долгосрочной токсичности для многих из многих методов лечения, которые были тщательно протестированы на моделях мышей. Наконец, мыши не могут быть лучшей моделью заболевания. В случае МДД было бы очень интересно увидеть результаты использования Debio-025 в модели собак, мышечной дистрофии золотистого ретривера. У собак GRMD болезнь более тяжелая, чем у мышей MDX, с заметным снижением подвижности и преждевременной смертью. Это происходит в течение первого года жизни собаки и поэтому позволяет клинически оценить пользу терапевтического подхода.
Несмотря на эти оговорки, уменьшение митохондриально-зависимого некроза является важным дополнением к ряду потенциальных способов лечения МДД и связанных с ним мышечных дистрофий. Вполне вероятно, что ни одного лечения не будет достаточно, чтобы полностью остановить или обратить вспять прогрессирование этих заболеваний, но комбинация методов лечения может однажды дать ответ на эти разрушительные условия.
Читайте также:
