
Генетические заболевания у детей мышцы
Существует огромное количество различных заболеваний, которые возникают у деток независимо от обстоятельств или действия окружающей среды. Это категория именно наследственных болезней. Сейчас же пойдет речь о такой проблеме, как мышечная дистрофия Дюшенна: что это за хворь такая, каковы у нее симптомы и можно ли с ней справиться.
Терминология
Изначально нужно узнать, что же такое наследственные болезни. Так, это заболевания, которые возникают в результате дефектов аппарата наследственных клеток. То есть это определенные сбои, которые происходят на генетическом уровне.
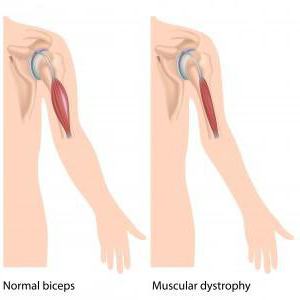
Мышечная дистрофия Дюшенна – это именно наследственная болезнь. Проявляется она очень быстро, основной симптом в данном случае – это быстро прогрессирующая слабость в мышцах. Нужно отметить: как и все остальные мышечные заболевания, болезнь Дюшенна также приводит в конечном результате к атрофии мышц, нарушению моторики и, конечно же, инвалидности. В подростковом возрасте детки с таким диагнозом уже не имеют возможности самостоятельно передвигаться и не могут обходиться без посторонней помощи.
Что происходит на генном уровне
Как уже было отмечено, мышечная дистрофия Дюшенна – это генетическое заболевание. Так, происходит мутация в том гене, который отвечает за выработку особого белка дистрофина. Именно он и необходим для нормальной работы мышечных волокон. При этом важно отметить, что эта генетическая мутация может как передаваться по наследству, так и возникать спонтанно.
Также важно отметить, что ген локализируется в хромосоме Х. Но женщины этой болезнью заболеть не могут, являясь только лишь передатчиком мутации от поколения к поколению. То есть если мама передаст мутацию сыну, он с 50%-й вероятностью заболеет. Если же девочке, она просто будет носителем гена, клинических проявлений болезни у нее не будет.
Симптоматика: группы
В основном, болезнь активно заявляет о себе примерно в 5-6 летнем возрасте. Однако первые симптомы могут возникнуть у малыша, который еще не достиг трехлетнего возраста. При этом надо отметить, что все патологические нарушения медки условно разделяют на несколько больших групп:
- Поражение мускулатуры.
- Поражение сердечной мышцы.
- Деформация скелета ребенка.
- Различные эндокринные расстройства.
- Нарушения нормальной умственной деятельности.
Наиболее часто встречающиеся проявления болезни
Обязательно также надо рассказать о том, как проявляется синдром Дюшенна. Симптомы бывают следующие:
- Слабость. Которая постепенно нарастает, развивается.
- Начинается прогрессирующая мышечная слабость именно с верхних конечностей, далее затрагиваются ноги и только потом – все остальные части тела и органы.
- Ребенок утрачивает возможность сам передвигаться. Примерно к 12-летнему возрасту такие детки уже полностью зависимы от инвалидной коляски.
- Также наблюдаются расстройства дыхательной системы.
- Ну и, конечно же, бывают нарушения в работе кардиологической системы. Позже происходят необратимые изменения в миокарде.
О поражении мышц скелета
Именно поражения мышечной ткани – наиболее распространенный симптом, если речь идет о такой проблеме, как синдром Дюшенна. При этом надо отметить, что рождаются детки без особых отклонений в развитии. В малом возрасте ребята менее активны и подвижны, нежели сверстники. Но чаще всего это связывают с темпераментом и характером ребенка. Поэтому отклонения очень редко замечаются. Более существенные признаки проявляются уже во время ходьбы малыша. Такие детки могут передвигаться на носочках, не становясь на полную стопу. Также они частенько падают.
Особым показателем является еще и симптом Говерса. То есть ребенок, чтобы подняться с пола, активно пользуется руками, как бы взбираясь по самому себе.
Также надо отметить, что при такой проблеме, как синдром Дюшенна, у ребенка постепенно атрофируются мышцы. Но нередко бывает так, что у крохи внешне мускулатура кажется очень развитой. Мальчик даже на первую вскидку оказывается как бы накачанным. Но это только лишь обман зрения. Все дело в том, что в процессе болезни мышечные волокна постепенно распадаются, а их место занимает жировая ткань. Отсюда и такой внушительный внешний вид.

Немного о деформации скелета
Если у ребенка прогрессирующая мышечная дистрофия Дюшенна, то постепенно у мальчика изменится форма скелета. Сначала патология затронет поясничный отдел, далее возникнет сколиоз, то есть произойдет искривление грудного отдела позвоночника. Позже проявится сутулость и, конечно же, изменится нормальная форма стопы. Вся данная симптоматика еще в большей степени будет сопутствовать ухудшению двигательной активности малыша.
О сердечной мышце
Обязательным симптомом при данной болезни также является поражение сердечной мышцы. Происходит нарушение ритма сердца, возникают регулярные перепады артериального давления. При этом сердце увеличивается в размерах. Но его функциональные возможности наоборот, уменьшаются. И в результате постепенно формируется сердечная недостаточность. Если эта проблема еще будет сочетаться с дыхательной недостаточностью, то возникает большая вероятность летального исхода.

Нарушения умственной активности
Нужно отметить, что мышечная дистрофия Дюшенна-Беккера не всегда проявляется таким симптомом, как умственная отсталость. Связано это может быть с дефицитом такого вещества, как аподистрофин, необходимого для работы головного мозга. Нарушения интеллекта могут быть самыми разными – от слабой умственной отсталости до идиотии. Усугублению этих когнитивных расстройств способствует еще и невозможность посещать садики, школы, кружки и иные места скопления детей. В результате возникает социальная дезадаптация.
Расстройства работы эндокринной системы
Различные эндокринные расстройства встречаются не более чем у 30-50% всех больных. Чаще всего это именно лишний вес, ожирение. При этом детки также имеют более низкий, чем у сверстников, рост.
Исход болезни
Какова клинико-эпидемиологическая характеристика мышечной дистрофии Дюшенна? Так, частота возникновения болезни – 3,3 пациента на 100 тысяч здоровых людей. Нужно отметить, что мышечная атрофия постепенно прогрессирует, и к 15-летнему возрасту мальчик уже не может обходиться без помощи окружающих, являясь полностью обездвиженным. Ко всему, происходит еще и частое присоединение различных бактериальных инфекций (чаще всего именно мочеполовой и дыхательной систем), при неправильном уходе за ребенком возникают пролежни. Если проблемы с дыхательной системой соединяются с сердечной недостаточностью, это грозит смертельным исходом. Если же говорить в общем, то такие пациенты практически никогда не живут более 30 лет.
Диагностика болезни
- Генетическое тестирование, то есть анализ ДНК.
- Электромиография, когда подтверждается первичное изменение мышц.
- Биопсия мышц, когда происходит определение наличия белка дистрофина в мышце.
- Анализ крови на определение уровня креатинкиназы. Нужно отметить, что именно этот фермент указывает на гибель мышечных волокон.
Лечение
Полностью излечиться от данной болезни невозможно. Можно только облегчить проявление симптомов, что сделает жизнь больного немного проще и удобнее. Так, после того, как пациенту ставят такой диагноз, чаще всего ему назначают терапию глюкокортикостеродами, которые призваны замедлить процесс развития болезни. Иные процедуры, которые также могут быть использованы при данной проблеме:
- Дополнительная вентиляция легких.
- Терапия медикаментами, которая направлена на нормализацию работы сердечной мышцы.
- Использование различных приспособлений, которые повышают мобильность пациента.
Также важно отметить, что сегодня ведутся разработки новейших методик, которые основаны на генной терапии, а также пересадке стволовых клеток.

Иные мышечные заболевания
Существуют еще и иные мышечные врожденные заболевания детей. К таким болезням можно отнести, помимо дистрофии Дюшенна:
- Дистрофию Беккера. Эта болезнь очень похожа на синдром Дюшенна.
- Мышечную дистрофию Дрейфуса. Это медленно прогрессирующая болезнь, при которой интеллект сохраняется.
- Прогрессирующую мышечную дистрофию Эрба-Рота. Проявляется в подростковом возрасте, прогрессирование быстрое, инвалидизация наступает рано.
- Плечелопаточно-лицевую форму Ландузи-Дежерина, когда мышечная слабость локализируется в области лица, плеч.
При этом надо отметить, что ни при одной из этих болезней не проявляется мышечная слабость у новорожденных. Вся симптоматика возникает в основном в подростковом возрасте. Длительность жизни пациентов чаще всего не превышает 30 лет.
Синдром Дауна и другие генетические заболевания у детей: какая коррекционная помощь нужна
В последние годы сильно возросло количество генетических нарушений у детей. Эту печальную тенденцию видит на своих консультациях и Наталья Керре — дефектолог, семейный консультант, автор книги "Особенные дети: Как подарить счастливую жизнь ребенку с отклонениями в развитии". Она описала самые часто встречающиеся в ее практике генетические синдромы — те, с которыми с наибольшей долей вероятности могут столкнуться родители. И рассказала, в чем может заключаться коррекционная помощь детям.

Генетика как наука пока только развивается, мы знаем о генетических аномалиях не очень много, но правильная и своевременная диагностика крайне важна для выбора педагогического и медицинского маршрута помощи ребёнку. Генетические синдромы могут принимать самый разный облик и быть похожи на умственную отсталость, аутизм, шизофрению, ДЦП.
Родителей должны насторожить два момента: если у ребёнка имеются аномалии физического облика (необычная форма ушей, пальцев, глаз, странная походка и т.д.) — и если специалисты долго не могут определиться с диагнозом (каждый ставит своё, пройдено уже больше пяти консультаций, но единого мнения нет).
От рождения ребёнка с генетическими проблемами не застрахована ни одна семья, но считается, что в зоне повышенного риска находятся следующие категории:
- Семьи, в которых уже есть ребёнок с любыми генетическими отклонениями.
- Мать старше 40 лет.
- В анамнезе есть самопроизвольные выкидыши либо замершая беременность.
- Длительный контакт родителей с мутагенными вредностями (облучение радиацией, "вредное" химическое производство и т.д.).
Рассмотрим наиболее часто встречающиеся генетические синдромы. Необходимо напомнить, что окончательный вывод по поводу диагноза делается только после очной консультации врача-генетика и всестороннего обследования ребёнка!
Синдром Дауна
Это наиболее изученное на сегодняшний день генетическое заболевание. У детей наблюдается снижение мышечного тонуса, недостаточно развитая моторика, нарушение функции вестибулярного аппарата. Синдрому Дауна также свойственны уплощённое лицо и затылок, низко расположенные уши, увеличенный язык, "монголоидный" разрез глаз. Однако эти физические особенности могут проявляться в разной степени. И, вопреки распространённому мнению, дети с синдромом Дауна довольно сильно отличаются друг от друга и больше похожи на своих родителей, чем друг на друга.
Эти дети обычно ласковые, артистичные, общительные, не склонные к антисоциальным поступкам. У детей может быть различный уровень снижения интеллекта: от глубокой умственной отсталости до незначительной задержки развития. Большинство детей способны к обучению и социализации по программе для лиц со снижением интеллекта.

Синдром Ретта
Это генетическое заболевание встречается только у девочек. Беременность и роды обычно протекают без проблем, новорожденные ничем не отличаются от других детей. Однако после 1,5–2 лет наступает регресс, когда ребёнок перестаёт осваивать новые навыки, снижаются темпы роста окружности головы.
Со временем добавляются дополнительные признаки: характерные "моющие" движения руками в области пояса, эпилептические приступы, остановки дыхания во сне, неадекватный смех и вскрикивания, замедление роста кистей рук, стоп и головы. Развитие идёт неравномерно, периоды остановки и регресса сменяются движением вперёд.
Уровень интеллектуального отставания различен, очень хорошие результаты при работе с детьми с синдромом Ретта даёт сочетание методик для детей с ДЦП с методиками для детей с аутизмом. Периоды регресса, конечно, существенно осложняют и замедляют коррекционную работу, но со временем она всё равно обязательно приносит свои плоды.
Синдром Мартина-Белл
Его еще называют синдромом ломкой Х-хромосомы: у детей большой лоб, низко посаженные оттопыренные уши при недоразвитии средней части лица. Рост небольшой, обычно есть снижение мышечного тонуса, косоглазие. Кожа бледная, очень хорошо растяжимая. Дети очень подвижные, эмоционально неустойчивые (возможен внезапный переход от смеха к слезам и обратно), тревожные.
Часто встречаются черты, похожие на аутизм: эхолалия, двигательные стереотипии, трудности с установлением глазного контакта, повышенная чувствительность к свету, звуку, прикосновениям. Почти у всех детей речевые проблемы: нарушение слоговой структуры слова, проблемы с артикуляцией, своеобразный назальный оттенок голоса и т.д.
Дети обычно хорошо подаются коррекции, охотно занимаются. Хорошие результаты показало использование сочетания методик для детей с аутизмом и снижением интеллекта.
Синдром Прадера-Вилли
При этом генетическом синдроме в возрасте 2-6 лет у детей появляется характерная особенность — аномально повышенный аппетит, отсутствие чувства насыщения. У детей с синдромом Прадера-Вилли наблюдается снижение мышечного тонуса, удлинённая форма головы, широкое плоское лицо, миндалевидные глаза, косоглазие, подковообразная форма рта.
Дети обычно эмоциональные, жизнерадостные, но после 6 лет может появиться психопатоподобное поведение с бурными истериками. Со временем повышается общая тревожность, наблюдается компульсивное поведение в виде "щипков" себя за кожу.
Почти у всех детей с синдромом Прадера-Вилли снижен интеллект, но часто очень хорошо развито визуальное восприятие. Дети хорошо обучаемы по программам для детей со снижением интеллекта, обычно легко учатся читать по методикам с применением глобального чтения.
Синдром Ангельмана
Характерный признак этого генетического заболевания — приступы беспричинного смеха, эйфории, застывшее на лице счастливое выражение. Дети гиперактивны, у них нарушена координация движений, часто тремор конечностей. У детей с этим синдромом, как правило, либо полностью отсутствует речь, либо присутствует 5-10 слов.
У детей наблюдается гипопигментация кожи, увеличение интервала между зубами, гладкие ладони, постоянная жажда, слюнотечение. Дети обычно мало и плохо спят. Часто — эпилептические приступы. Интеллект снижен. Хорошие результаты даёт применение сочетания методик для детей с интеллектуальной недостаточностью с методиками для детей с гиперактивностью.
Родителям необходимо помнить, что постановка ребёнку диагноза, связанного с генетическими аномалиями, не означает, что коррекционная работа будет бессмысленной. К сожалению, на сегодняшней день не существует способа полностью вылечить генетический синдром. Но улучшить состояние ребёнка по сравнению с изначальным можно абсолютно во всех случаях.

Мышечная дистрофия, или миопатия Дюшенна, — тяжелая наследственная патология, которая постоянно прогрессирует. Замедлить мышечное разрушение практически невозможно.
Связано это с врожденными изменениями. Впервые о миопатии Дюшенна заговорили в середине XIX века. Обнаружил эту патологию французский невролог. В тот момент был известен один тип течения болезни, через некоторое время выделили еще несколько способов развития состояния.
Этот тип болезни сильно похож на миодистрофию Беккера, но в то же время отличается от него сложностью и внешними признаками.
Миодистрофия Дюшенна обнаруживает у 1 ребенка из 4000. Этот тип патологии относится к самым распространенным мышечным дистрофиям, относится к врожденным заболеваниям.
Этиология нарушения
Одному из генов в структуре генома человека присвоили имя невролога, в честь которого и было названо отклонение. На мышечную дистрофию Дюшенна могут влиять разные факторы:
- кровосмешение;
- предрасположенность генетического характера, например, при наличии миопатии Дюшенна у кого-либо из родни;
- неправильный синтез мышечных волокон, ускоренное распространение и замещение жировой прослойкой, соединительными волокнами;
- наследственные формы синдрома Дюшенна, чаще всего переходящие от матери;
- мутация генома при формировании во время беременности;
- аномалии в хромосомных структурах неясного происхождения;
- сильные нарушения в развитии дистрофина;
- патологические изменения биохимии в крови.

Характеристика наследственной патологии
Генетическая природа заболевания была сразу же доказана после обнаружения синдрома в 1868 году. Эта патология почти идентична с миодистрофией Беккера, то есть, обладает теми же генетическими предпосылками для формирования.
Однако миодистрофия Беккера отличается иными симптомами. Для болезни характерны следующие особенности:
- диагностируется у мальчиков до 5 лет;
- прогрессирует стремительно;
- у девочек никогда не обнаруживается;
- атрофия мышц обладает ступенчатым развитием – сначала страдает тазовый пояс;
- затем вовлекаются мышцы ног;
- после этого миопатия Дюшенна поражает мышцы спины, плеч;
- завершается прогрессирующая мышечная дистрофия Дюшенна поражением рук;
- специфический признак нарушения – деформация позвоночника, чаще встречающаяся в форме кифоза или лордоза;
- миодистрофия Дюшенна почти всегда сопровождается повреждениями грудины и стоп, они становятся неправильной формы, сильно меняют тело человека;
- при патологии, в отличие от миодистрофии Беккера, появляется повреждение левого сердечного желудочка, аритмия и кардиопатия;
- примерно у 30% пациентов развивается олигофрения.

Мышечная дистрофия Дюшенна никогда не протекает в легкой степени, всегда имеет крайне неблагоприятный прогноз. Развивается быстро, возможность ходить пациент утрачивает уже к 12 годам. При мышечной дистрофии Дюшенна смерть наступает из-за инфекции бронхов или легких, после остановки сердца.
Симптомы нарушения
Первые признаки миопатии Дюшенна встречаются уже в возрасте 1,5 лет. В редких случаях их не удается заметить до 5 лет. Проявляются признаки заболевания Дюшенна сначала в легкой степени. Их комбинация зависит от общего состояния здоровья:

На фоне мышечной дистрофии Дюшенна у маленького пациента развивается острая депрессия, которую дети с трудом переносят. Нередко причиной смертности при миодистрофии Беккера и Дюшенна становится суицид.
Диагностика заболевания
Мышечная дистрофия Дюшенна крайне тяжело поддается диагностики. Для этого привлекают комплекс методов. Первое, что нужно пройти при подозрении на миопатию Дюшенна, — это ЭКГ. Для подтверждения диагноза необходимо, чтобы анализ показал нарушения стенки левого желудочка.

Следующий этап – это определение уровня дистрофина, который не меняется в сторону обычной дистрофии. Также необходимо сдать кровь на биохимический анализ. Если есть миодистрофия Беккера или болезнь, названная в честь французского невролога, отмечается высокий уровень КФК.

Тактика лечения заболевания
Чтобы лечение мышечной дистрофии Дюшенна было эффективным, нужно четко следовать намеченному плану после постановки диагноза. Излечению болезнь никогда не поддается полностью, но можно значительно облегчить жизнь пациента. Современная медицина способна замедлить миопатию Дюшенна следующими методами:

Важно! Поддержать здоровье при мышечной дистрофии Дюшенна можно и другими методами – ЛФК и электрофорезом.

В тяжелых случаях всё лечение проводят в домашних условиях, если есть медицинские возможности для организации сложной терапии специальными приборами.
Обязательное условие для лечения миопатии Дюшенна – постоянное наблюдение у кардиолога. Также необходимо составить грамотное меню. При заболевании нужно есть много овощей, приготовленных на пару, фруктов, растительных жиров и нежирного мяса. Запрещено употребление алкоголя, кофеина и крепкого чая.
Последствия и осложнения
В 100% случаев миопатия Дюшенна сопровождается тяжелыми последствиями для организма и сильно укорачивает жизнь. Пациент всегда умирает от осложнений заболевания – остановки сердца или легочной инфекции.
Если мышечную дистрофию Дюшенна удалось обнаружить в раннем возрасте, есть шанс, что человек доживет до 30 лет. Но только при условии адекватной терапии и комплексного подхода. Среди осложнений миопатии Дюшена нередко выделяют остеопороз, поражения позвоночника и суставов, а также патологии пищеварительной системы.

Нервно-мышечные заболевания (НМЗ) – что это такое?
В медицине под этим термином подразумевается большая группа болезней, передающихся на генетическом уровне от родителей к ребенку.
Им характерно нарушение функций мускулатуры, низкая двигательная активность или ее отсутствие.
Такие последствия наступают в результате нарушений функций нервно-мышечных соединений, поражения мышц, спинномозговых нейронов или нервов.
Основные причины возникновения НМЗ – это аутоиммунные болезни, отравление различного рода веществами и наследственный фактор. Также сюда стоит отнести часть врожденных дефектов метаболизма.
Классификация
К НМЗ относятся:
- первичные прогрессирующие мышечные дистрофии (миопатии);
- вторичные прогрессирующие мышечные дистрофии;
- врожденные непрогрессирующие миопатии;
- миотонии;
- наследственныепароксизмальные миоплегии.
О каждом из видов поговорим подробнее дальше.
Первичные прогрессирующие мышечные дистрофии (миопатии)

К признакам прогрессирующих мышечных дистрофий (ПМД) относятся дегенеративные изменения в мышцах.
Они заключаются в истончении мускулатуры, замене мышечной ткани на жировую и соединительную.
Возникает фокальный некроз, поперечная исчерченность теряется.
Некоторые механизмы возникновения и течения болезни до сих пор не установлены. Причиной миопатии являются мембранные дефекты в мышечных клетках. Большие надежды на установление деталей патогенеза возлагают на микробиологов.
При миопатиях мышцы слабеют, затем атрофируются. Существует несколько форм этой болезни. Разные виды ПМД различают по типу наследования, срокам проявления, характеру и течению болезни, расположением атрофий.
Вторичные прогрессирующие мышечные дистрофии
При вторичных дистрофиях (спиральных и невральных амиотрофиях) поражаются спинномозговые мононейроны и периферические двигательные нервы. Это приводит к вторичному поражению мышц и их атрофии.
Болезни этого типа прогрессируют. При изучении мышц больного обнаруживаются как нормальная, так атрофированная и гипертрофированная мышечная ткань.
Наследственные пароксизмальные миоплегии
Пароксизмальные миоплегии – редко встречающиеся наследственные заболевания у детей. Характеризуется периодическими приступами паралича мышц скелета. Наследуется по принципу, при котором для проявления болезни достаточно одного мутантного аллеля, локализованного в аутосоме.
Выделяют три формы:
- гиперкалиемическая;
- гипокалиемическая;
- нормокалиемическая.

Гипокалиемической формой пароксизмальной миоплегии чаще всего болеют мужчины.
Первые приступы заболевания появляются с 10 лет.
Приступ возникает в утренние или ночные часы. При этом чувствуется слабость в конечностях, шее, часто доходящая до паралича. В отдельных случаях паралич распространяется на лицевую мускулатуру и дыхательные пути.
Приступ сопровождается жаждой гиперемией, потливостью. Длительность может составлять от часа до семи дней. Женщины обычно испытывают приступ в первый день менструаций.
Гиперкалиемическую форму можно встретить реже, ее приступы начинаются в младшем возрасте. Приступ провоцируют холод и длительное бездействие мышц. Он начинается с расстройства чувствительности лицевых мышц, рук и ног.
Третья форма болезни проявляется до 10-летнего возраста. Приступы проходят в течение нескольких дней или недель. Они могут быть спровоцированы низкой температурой окружающей среды, употреблением алкоголя, большими физическими нагрузками.
Справка! Наследственные пароксизмальные миоплегии могут быть симптомами болезней щитовидной железы, Адиссона, реже встречаются при рвоте и поносе.
Врожденные непрогрессирующие миопатии
Непрогрессирующая миопатия характеризуется доброкачественностью, проявляется в младенчестве или в детстве. К этой разновидности НМЗ относятся болезни центрального стержня, намалиновые и митохондриальные миопатии. При этом возникают метаболические мышечные изменения. Пациент чувствует снижение сил, ослабление рефлексов.
Миотонии

Миотония – это замедленное расслабление мышцы после ее напряжения.
Болезнь разделяют на три вида:
- миотония действия;
- перкуссионная миотония;
- электромиографическая миотония.
Если больной с миотонией действия сожмет пальцы в кулак, а затем попытается быстро их разогнуть, то ему понадобится время, чтобы полностью выпрямить ладонь.
Вторая разновидность отличается сокращением мышц при интенсивном постукивании молоточком.
Чтобы обнаружить электромиографическую миотонию, потребуется игольчатый разряд. При этом аппарат показывает разряды высокой частоты, которые сначала учащенные, а потом снижают частоту.
Список всех НМЗ
- Болезни ЦС;
- Миотонии Томсена;
- Болезни Штейнерта-Куршманна;
- Миодистрофии Беккера;
- Миодистрофии Дюшенна;
- Миодистрофии Ландузи-Дежерина;
- Митохондриальные миопатии;
- Невральные амиотрофии Шарко-Мари-Тута;
- Немалиновые миопатии;
- Офталъмоплегические миопатии;
- Спинальные амиотрофии Верднига-Гоффманна;
- Спинальные амиотрофии Кугельберга-Веландер;
- Врожденная мышечная дистрофия;
- Мышечная дистрофия Беккера;
- Болезнь Помпе;
- Миотоническая дистрофия;
- Мышечная дистрофия Дюшенна.
Симптомы
Определяющий симптом НМЗ – это слабость в мышцах. Зонами поражения являются плечевой пояс, бедра, таз, область плеч. При каждом виде болезни поражается конкретная группа мышц. Мышцы поражаются симметрично.
Больной задействует другие мышцы, чтобы справиться с определенной задачей. Например, если поражена область ног, то, чтобы подняться с пола, человек сначала опирается руками, встает на колени, берется за опору, а потом уже может сесть на кресло. Сесть, не используя руки, он не может.
Справка! Редко встречаются случаи функциональных нарушений лицевых мышц. При этом возникает опущение века, губы, появляются речевые нарушения, трудно глотать.
Большинство заболеваний проходят с одинаковыми признаками. Со временем мышца атрофируется, возникает псевдогипертрофия из-за разрастания соединительной ткани. Возникает боль, подвижность в суставе ограничивается.
Диагностика
В диагностике нервно-мышечных заболеваний важны следующие методы:
- биохимические;
- электрофизиологические;
- патоморфологические;
- ДНК-диагностики.
Биохимический метод заключаются в том, что у больного определяют ферменты мышц.
Важно! Повышение уровня ферментов может быть вызвано и у здоровых людей при физической нагрузке.
При разных видах НПЗ уровень фермента повышается в разное количество раз по сравнению с нормой. Это позволяет определить форму заболевания.
Электрофизиологический метод включает в себя электронейромиографию в сочетании с электромиографией.
Патоморфологический метод включает мышечную биопсию – изучение мышцы под световым микроскопом.
Наиболее эффективным методом определения болезни является ДНК-диагностика. Полимеразная реакция позволяет выявить НПЗ в 70% случаев.
Способы лечения

Лечение направлено на поддержку мышечных сил, замедление атрофирования.
Цель терапии заключается в том, чтобы пациент как можно дольше передвигался без посторонней помощи, т.к. в постоянном вертикальном положении возникают расстройства дыхания.
Основными способами лечения являются:
- Лечебно-физкультурный комплекс. Это различные активные и пассивные движения. Нельзя подвергать больного усиленной нагрузке. Упражнения должны быть регулярными;
- Ортопедические мероприятия. Применение специальных шин и проведение операций. Эти меры направлены на сохранение самостоятельных передвижений;
- Лекарственные препараты. Медикаменты, поддерживающие обмен веществ, устраняющие дефицит энергии и белка. Больным назначают фосфаден, преднизолон, нифедипин, витамин Е.
Неправильно поставленный предварительный диагноз приводит к ошибкам в диагностике формы НМЗ. Это увеличивает траты на осуществление дорогостоящих анализов и препятствует осуществлению профилактики рецидива, определения генетического статуса предков пациента и подбора адекватного патогенетического лечения, которым можно ограничиться в некоторых случаях.
Полезное видео по теме:
Читайте также:
