
Спинально мышечная атрофия у детей истории
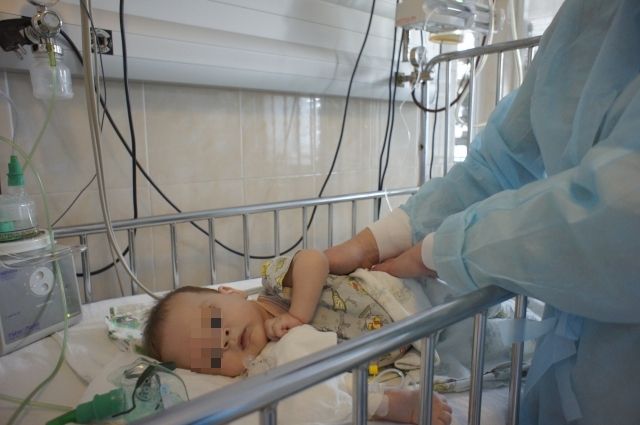
Младенец — надежда детей со СМА
У 8-месячного Саши Лабутина генетическое заболевание: спинальная мышечная атрофия. Эта болезнь не влияет на интеллект, но постепенно лишает человека возможности двигаться, а потом — самостоятельно глотать и дышать. В зависимости от тяжести заболевания дети с СМА теряют возможность двигаться самостоятельно к 18 годам (при легкой степени) или к 2 годам (при самой тяжелой форме, когда дети даже сидеть не могут, поэтому о ходьбе речи нет).
В большинстве случаев родители, являясь носителями этого генетического заболевания, не знают об этом. В России нет массового скрининга на СМА во время беременности и на этапе планирования. Он проводится только по 5 генетическим заболеваниям. А их — тысячи.

У Саши тяжелая степень заболевания, он даже не успел научиться переворачиваться и ползать.
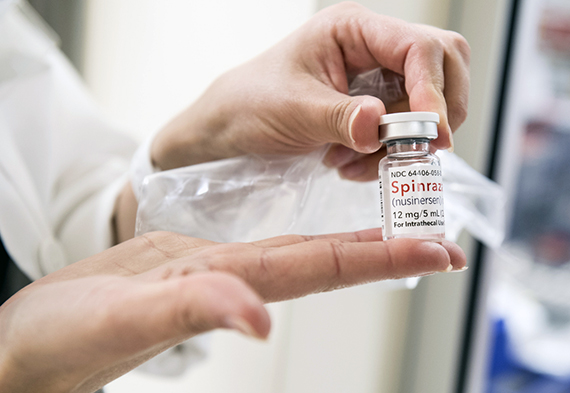
Саша — надежда многих пациентов с таким же диагнозом. Он участвует в испытаниях препарата, который используется в лечении СМА. Если они пройдут успешно, его разрешат к использованию в нашей стране. Сейчас российским пациентам прописывают только поддерживающие препараты. В среднем ребёнок принимает по 4-5 препаратов, но ни один из них не лечит.
Появилась надежда?

Как живут дети с СМА
У 15-летнего Миши Ерина СМА 3 типа, он даже мог ходить в школу до 4 класса. Затем пришлось перейти на домашнее обучение. Бабушка Валентина Арсентьевна вспоминает, что, учась в школе, Миша очень любил петь и на уроках музыки был запевалой.
Сейчас он увлекается IT и программированием, очень любит метеорологию. Ему даже подарили на день рождения экскурсию на университетскую метеорологическую станцию. Пока посетить не удалось, и в ближайшее время не получится. Мише нужно досрочно сдать все экзамены и контрольные работы за 8 класс. В мае мальчику нужно ехать в Федеральный центр им. Илизарова в Кургане, где ему предстоит операция на позвоночнике, чтобы исправить сколиоз.
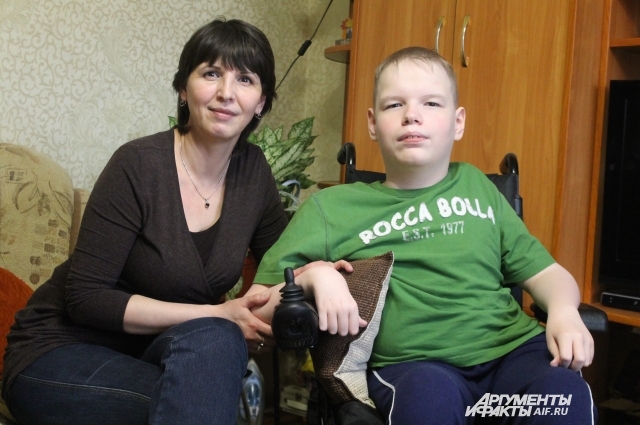

Из-за того, что заболевание редкое, не все врачи знают, как лечить, какой подход нужен к ребёнку и на что важно обратить внимание. В случае с Мишей это фатальным не стало, но упущения в лечении уже есть.
Она отмечает, что в столичных больницах и поликлиниках специалисты более осведомлены. Когда выявляется пациент с СМА, за его здоровьем следит мультидисциплинарная бригада врачей, состоящая не только из невролога и ортопеда, как это бывает в регионах. В неё входят также пульмонолог, кардиолог, терапевт или педиатр, реабилитолог и другие специалисты, которые принимают решение о лечении и реабилитации коллегиально.
Но детям нужна не только медицинская помощь.

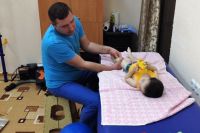
Теперь Миша дважды в неделю вместе с бабушкой ездит на занятия. Он старается не пропускать ни дня.
Старший брат Миши — тоже программист. Он уже окончил университет и сейчас работает по специальности. Бабушка Валентина Арсентьевна рассказывает, что внуки часто устраивают споры на компьютерные темы.
Мария Баженова рассказывает, что мальчика на следующий учебный год хотят взять в проектную группу одарённых детей, настолько его результаты превосходны.

Глоток воздуха
Если для Саши Лабутина и тех детей с СМА, кто родится в будущем, есть крохотная надежда на излечение, то для взрослых пациентов остаётся только поддерживающая терапия и специальное оборудование.
Как правило, детям с СМА обязательно требуется дыхательный мешок или мешок Амбу, чтобы проводить дыхательную гимнастику и поддерживать лёгкие, откашливатели. Большинство семей обращаются в благотворительные фонды за помощью, потому что в среднем стоимость необходимого оборудования для ребёнка с СМА — от 1 млн рублей. Также требуются расходные материалы к оборудованию стоимостью порядка 500 тысяч рублей в год.
В семье работают и мама, и папа, и старший брат Миши, но сумма, которая требуется, для них неподъемна: 965 тысяч рублей. А оборудование очень важно. Без откашливателя любая простуда может обернуться для мальчика воспалением лёгких. Кровать поможет Мише самостоятельно переворачиваться и ложиться. Когда он был младше, родители и бабушка помогали ему, но Миша подрос и мама с бабушкой уже не справляются, а папа работает сутки через двое.
Вихрастый мальчишка задумчиво смотрит с фото в соцсетях. Через три месяца Захару Рукосуеву из Красноярского края должно было исполниться три года, но… 30 января ребенок умер в реанимации краевой детской больницы. У него было редкое заболевание - спинальная мышечная атрофия.
Помощь не пришла
Доза ценою в жизнь
Потерявшие ребенка родители готовы идти в суд. Чтобы добиться справедливости, помочь другому сыну и таким, как он.
Не успели…
А что же говорят в минздраве ? СМА – заболевание неизлечимое, констатируют представители ведомства. Оно приводит к поражению нервной системы, постепенной атрофии мышц и летальному исходу.
В Красноярском крае 16 пациентов с СМА. Ведется работа по возможности включения краевых пациентов в Программы, действующие на территории России. Главным внештатным детским неврологом минздрава края проводится подбор детей для участия в программе.

Кроме того, министерством здравоохранения вместе с представителями законодательной власти обсуждается вопрос по привлечению к сотрудничеству благотворительных фондов. Планируется выйти с этим предложением для переговоров к представителям уже работающих на территории благотворительных организаций, либо обсудить вопрос специализированного фонда для таких пациентов.
Значит, все это вопрос времени. Увы, его у Захара как раз и не было…
- Малыш, прости этот мир, - пишут в соцсетях. – Не успели…
А В ЭТО ВРЕМЯ
ДАЙТЕ ШАНС!
Среди 16 человек с диагнозом СМА в Красноярском крае – 5-летняя Анжелика Сазонова . У девочки мышечная атрофия второго типа, ходить сама она не может.
«Каждый наш день - это борьба с болезнью, постоянные занятия, реабилитации, ведь если не будет физиопроцедур и нагрузки (ЛФК, бассейн, пассивная гимнастика, массаж), развитие болезни пойдет колоссальными темпами, - рассказывает мама малышки. - Ежедневная реабилитация, к сожалению, не излечивает заболевание, а лишь замедляет процесс прогрессирования.
Такой мальчуган крутой, и теперь он не в нашем мире. У меня мурашки по коже. Его мама, так же, как и я, обращалась в минздрав, получила отказ, и малыш умер. Светлая память Захарчику.

Но его брату, другим детям с СМА, нужна помощь. Важен каждый рубль. Возможно, это даст ребенку еще один шанс – на жизнь без мучений. Как помочь Анжелике? Можно посмотреть здесь.

Анжелика Сазонова. Фото: соцсети
Есть несколько типов спинальной мышечной атрофии: если одни пациенты умирают в младенчестве, то другие могут прожить до пожилого возраста. Кстати, у больных с СМА интеллектуальный уровень зачастую выше среднего.
СМА I - типа обнаруживается в раннем возрасте, стремительно прогрессирует. Это самая тяжелая форма, ребенок не может даже сам сидеть. Без респираторной поддержки не доживает до двух лет.
СМА II - типа тоже проявляет себя в детстве. Но развивается не так быстро. Ребенок может сидеть, но не способен сам ходить.
СМА III - типа диагностируют позже. К тому времени пациент может сам передвигаться, но постепенно теряет эту способность. Человеку приходится пользоваться инвалидной коляской.
Также существуют формы спинальной мышечной атрофии и у взрослых.
КОММЕНТАРИЙ
Андрей Носырев, главный детский невролог Красноярского края:
- К сожалению, при диагностике СМ есть проблемы. В том смысле, что, к примеру, синдром Дауна, выявляется на ранних сроках беременности, а СМА либо на позднем сроке беременности (и то не всегда), либо по факту, когда ребенок уже родился. Это может быть 6-й месяц после рождения. Диагностика орфанных заболеваний при беременности зависит от направленности – не все они касаются неврологии, как СМА (есть эндокринные, ортопедические патологии). Иногда болезни реализуются на более позднем сроке, даже у взрослых людей.
Если в семье уже имеется ребенок с СМА, то вероятность проявления болезни при последующей беременности очень высокая – это в случае, если один родитель является носителем патологического гена. Если он имеется у обоих родителей, то вероятность стопроцентная. И понимание этой ситуации родителями отличается, конечно, от понимания генетиков или врачей.
Дети, больные СМА 1, получают комплексную симптоматическую терапию, направленную на устранение дыхательных нарушений и респираторную поддержку; профилактику костных деформаций (сколиоза и контрактур), профилактику осложнений. Для замедления прогрессирования заболевания применяются комплексные схемы лечение: нейрометаболиты, лекарства, улучшающие нервно-мышечную передачу, средства, улучшающие кровообращение.
ЧИТАЙТЕ ТАКЖЕ:
Жизнь стоит 8 миллионов. В Красноярском крае, не дождавшись лечения, умер мальчик с редкой болезнью
СМА — это болезнь, которая постепенно разрушает мышцы, не оставляя никаких шансов на выживание: сначала отказывают мышцы рук, потом ребенок перестает ходить, сидеть, дышать. (Подробности)

Частота заболевания
Причина СМА
СМА — наследственное заболевание, оно связано с мутациями в гене SMN1.
Чтобы болезнь проявилась, носителями мутации в этом гене должны быть оба родителя. Рецессивный ген СМА имеет примерно каждый 40-й. Вероятность рождения больного ребенка от двух носителей – 25%, с такой же вероятностью ребенок двух носителей не будет иметь генной поломки. Ещё в 50% случаев он будет носителем СМА, но сам не заболеет.
В редких случаях (менее 2%) больные дети рождаются в семьях, где носителем является только один родитель. У второго родителя мутация гена происходит при закладке яйцеклетки или сперматозоида.
Что повреждается в результате мутации
Из-за дефектного гена в организме нарушается выработка белка SMN — протеина выживаемости мотонейронов. Без этого белка мотонейроны – нервные клетки спинного мозга, отвечающие за координацию движений и мышечный тонус – отмирают, сигнал в мышцы ног, спины и отчасти рук не идёт.
Без необходимого тонуса мышцы постепенно атрофируются. Отсутствие мышц пресса и спины приводит, кроме прочего, к обширным искривлениям позвоночника, а они – к проблемам с дыханием, которые из-за слабых мышц и так есть.
Болезнь может проявляться с первых месяцев жизни или в более позднем возрасте.
От чего зависит степень тяжести болезни
За выработку белка SMN отвечают два гена — SMN1 и SMN2.
Копий SMN2 в геноме бывает до восьми. От имеющегося у человека числа копий SMN2 и зависит тяжесть состояния больного. Такой сложный механизм болезни приводит к тому, что СМА имеет несколько форм, и состояние больных – очень разное.
Какие формы СМА существуют?
Существует 4 типа СМА, различающиеся степенью тяжести и возрастом, в котором впервые проявляется заболевание.
СМА I, болезнь Верднига-Гоффмана. Самая тяжёлая форма болезни, проявляется у младенцев от 0 до 6 месяцев. Дети с этой формой с рождения имеют трудности с дыханием, сосанием и глотанием, а также не осваивают самые простые контролируемые движения — не держат голову, не сидят самостоятельно. Ранее считалось, что большинство (80%) не доживают до двух лет. Сейчас благодаря новым стратегиям ИВЛ и зондовому кормлению срок жизни можно продлить ещё на несколько месяцев.
СМА II, болезнь Дубовица. Первые проявления болезни в 7-18 месяцев. Человек с таким типом СМА может есть и сидеть, но не ходит самостоятельно. Продолжительность жизни зависит от степени поражения мышц, обеспечивающих дыхание.
СМА III, болезнь Кюгельберга-Веландер. Болезнь впервые проявляется после полутора лет. Такие больные могут стоять (испытывая боль), но не ходят. На продолжительность жизни СМА III типа, как правило, не влияет, но сильно ухудшает её качество.
СМА, связанные с нарушением гена SMN, в медицинской литературе называют проксимальными — они составляют 95% от всех спинальных амиотрофий. СМА, не связанных с геном SMN, довольно много, но встречаются они редко. К ним относится, например, болезнь Кеннеди. Исследования 1990-х годов показали, что болезнь Кеннеди не связана не с поломкой гена SMN1, но с другими генетическими мутациями, приводящими к нарушению усвоения белка SMN. Болезнь проявляется у людей старше 35 лет. Для СМБА характерна, в основном, слабость конечностей.
Один из видов СМА, не связанный с геном SMN, называется болезнь Кеннеди. То, что эту болезнь до сих пор иногда относятся к СМА – анахронизм. В конце 1960-х, когда было выполнено подробное описание этой атрофии, её посчитали разновидностью СМА, так как при ней поражаются те же нервы и мышцы, что и при трёх типах СМА (но в гораздо меньшей степени).
Как это лечат?
На сегодняшний момент радикального лечения от СМА не существует.
Можно ли помочь больным СМА и как именно?
Вылечить болезнь пока нельзя, но можно облегчать состояние больных СМА, то есть различными способами компенсировать проявления болезни.
При тяжёлых типах СМА больным приходится помогать дышать и глотать. Поэтому им жизненно необходимы мобильные аппараты ИВЛ, аспираторы-откашливатели, мешки Амбу.
Ещё детям со СМА очень нужна помощь волонтёров, способных хоть на короткое время подменить родителей.
Детям, больным СМА, помощь может понадобиться в любой момент, поэтому мамы и папы всегда начеку и сами осваивают навыки реанимации, необходимые на случай, если ребенок внезапно перестал дышать.
Менее тяжёлым больным нужны лекарства, облегчающие дыхание, корсеты, коляски и другие приспособления, облегчающие перемещение и жизнь людей со слабыми мышцами.
Болезнь, продолжающаяся много лет, выматывает, поэтому пациентам, особенно взрослым, часто необходима помощь психолога.

Фонд работает по всей территории России. Работа фонда имеет два основных направления — оказание помощи самим больным СМА и их близким и работа на системные изменения ситуации со СМА в России.
Вы можете поддержать деятельность фонда, сделав пожертвование любым удобным для вас способом. Помочь можно, оформив разовое или регулярное пожертвование на специальной странице фонда или отправив на короткий номер 3443 смс со словом СМА и, через пробел, суммой пожертвования — например, СМА 300.
Можно ли заболеть СМА из-за прививок?
В Европе и США связь между прививками и проявлением болезни не прослежена.
Как определяют, что у ребенка именно СМА, а не какая-то другая болезнь?
Несмотря на то, что впервые СМА была описана австрийским неврологом Гвидо Верднигом и немецким неврологом Джоханном Хоффманном ещё в начале 1890-х годов, полностью понять природу заболевания удалось только в конце XX века. Ген SMN1 был открыт в 1995 году. Чтоб подтвердить диагноз СМА, нужен генетический тест.
В России соответствующие генетические тесты стали доступны в начале 2000-х годов. Генетический тест на СМА возможно сделать по ОМС, однако на практике не слишком много врачей знают этот редкий диагноз и направляют больных на соответствующее исследование. Стоимость такого тестирования в коммерческих лабораториях Москвы – порядка 6 тысяч рублей.
Сколько больных СМА в России?
Кто в России помогает людям со СМА и их семьям
Известные люди со СМА

Российская певица Юлия Самойлова родилась в городе Ухта (Республика Коми) В возрасте десять лет выступила на благотворительном концерте, после чего была приглашена заниматься пением в местный Дворец пионеров. В пятнадцати лет начала заниматься в городском Доме культуры.
Программист из Владимира Валерий Спиридонов. Окончил школу с золотой медалью, затем защитил диплом инженера. В 2015 году Валерий планировал стать участником эксперимента итальянского хирурга Серджио Канаверо по пересадке головы человека (эксперимент был отменен).

Что такое спинальные мышечные атрофии у детей. Спинальная мышечная атрофия у детей второго типа
Дебют заболевания начинается в возрасте от восьми до четырнадцати месяцев от рождения. Характерны генерализованная мышечная слабость, гипотония.
Истонченные пальцы, фасцикулярный тремор мышц проксимальных отделов конечностей, вытянутых кончиков пальцев, фибриллярные подергивания мышц.
Около двух лет у ребенка снижаются, затем исчезают сухожильные рефлексы конечностей. Также наблюдается слабость межреберных мышц и, как результат, уплощение грудной клетки, задержка двигательного развития. Лишь четвертой части больных детей удается самостоятельно сидеть или стоять с поддержкой. На поздних этапах данного заболевания у большинства детей диагностируется выраженный кифосколиоз. Течение этого заболевания также неблагоприятное, с летальным исходом в период с одного до четырех лет. Частой причиной летальных исходов является поражение дыхательных мышц, пневмония.
Это редкое аутосомно-рецессивное наследственное нервно-мышечное заболевание, вызванное прогрессивной дегенерацией клеток спинного мозга, которое приводит к прогрессирующей мышечной слабости.
В Германии, Италии, да и во многих других странах, все необходимое таким деткам предоставляет государство, это и оборудование и лекарства и всё всё, к сожалению в России такого нет, всё ложится на плечи родителей.

Спинальная мышечная атрофия (СМА или SMA) — разнородная группа наследственных заболеваний, протекающих с поражением и потерей моторных нейронов передних рогов спинного мозга.
Для спинальных мышечных атрофий Характерно нарушение работы поперечнополосатой мускулатуры нижних конечностей, а также головы и шеи. У больных отмечаются нарушения произвольных движений — ползание, ходьба, удержание головы, глотание. Мышцы верхних конечностей обычно не страдают. Для спинальных амиотрофий характерно сохранение чувствительности, а также отсутствие задержки психического развития.
История и патогенез.
Спинальная мышечная атрофия у детей впервые была описана G. Werdnig в 1891 году. G. Werdnig представил описание патоморфологических изменений различных групп мышц, периферических нервов и спинного мозга, отметив симметричную атрофию клеток передних рогов спинного мозга и передних корешков. В 1892 г. J. Hoffmann обосновал нозологическую самостоятельность заболевания. В дальнейшем G. Werdnig и J. Hoffmann (1893) доказали, что заболевание сопровождается дегенерацией клеток передних рогов спинного мозга. В 1956 г. Е. Kugelber^fa L. Welander выделили новую нозологическую форму спинальной мышечной атрофии, которая характеризуется более поздним началом и относительно доброкачественным течением по сравнению с описанной G. Werdnig и J. Hoffmann.
SMA вызвана мутацией в части ДНК, называемой SMN1 ген, который обычно производит белок SMN. Из-за мутации гена, у людей со SMA производится меньшее количество SMN белка, что приводит к потере моторных нейронов.
Классификация типов СМА.
• Тип 1, или болезнь Werdnig-Hoffmann, — наиболее неблагоприятная форма SMA.
Дети испытывают недостаток моторного развития, имеют трудности с дыханием, затруднения с сосанием и глотанием. Тип 1 SMA проявляется у детей в период от рождения до 6 месяцев.
• Тип 2 — несколько более благоприятнее.
Пациенты способны сидеть без поддержки или даже стоять с поддержкой и обычно не страдают при приёме пищи. Однако они имеют увеличенный риск осложнений от инфекций дыхательных путей. Тип 2 SMA проявляется у детей в период 7-18 месяцев.
Спинальная мышечная атрофия или СМА – генетически обусловленная патология, обнаруживаемая у младенцев, детей дошкольного возраста, подростков и взрослых и сопровождающаяся равносторонней атрофией нейронов спинномозговых передних рогов и корешков периферических нервов, что приводит к снижению мышечного тонуса и прогрессирующему параличу. В первом случае медики вынуждены констатировать тот факт, что ребенок никогда не сможет самостоятельно стоять, сидеть и ходить. В остальных он будет постепенно утрачивать эти способности и однажды окажется прикованным к инвалидному креслу.


Что такое спинальная мышечная атрофия и ее виды
Под этим термином объединяется несколько различных видов наследственных заболеваний, сопровождающихся ограничением двигательных способностей. Этим и объясняется тот факт, что в части случаев нарушения обнаруживаются не в младенческом возрасте, а у подростков или уже зрелых людей.
Впервые заболевание было описано в 1891 г. Г. Верднигом и в 1892 г. было выделено в отдельную нозологическую единицу Дж. Хоффманом, благодаря стараниям которых и получила свое второе название. Примерно через полвека Е. Кугелбергом и Л. Веландером была открыта другая подобная болезнь, развивающаяся в более позднем возрасте и отличающаяся более благоприятным течением.
Различают следующие формы патологии:
- СМА 0;
- СМА 1 (тяжелая форма);
- СМА 2 (промежуточная форма);
- СМА 3 (легкая форма);
- СМА 4 (поздняя форма).
Все их объединяет то, что причина их возникновения кроется в мутации рецессивного гена 5 хромосомы SMN. Это приводит к сбоям в продукции протеинов в организме, являющихся строительным материалом всех клеток. В результате страдают мотонейроны спинного мозга и постепенно разрушаются. Поскольку без них невозможна передача нервных импульсов к мышечным волокнам, они постепенно атрофируются, что становится причиной утраты способности двигаться.

К счастью, даже при наличии у обоих родителей мутации гена SMNу них с 75% вероятностью может родиться здоровый ребенок. Но практически всегда он также будет носителем этого гена. Поэтому при планировании беременности стоит проходить генетическое исследование, особенно при наличии случаев СМА в семье.
Это врожденная болезнь, признаки которой обнаруживаются обычно еще в роддоме. Она встречается редко и ее часто объединяются со СМА-1. Для этого вида типично абсолютное отсутствие подвижности, слабость мышц, отсутствие сухожильных рефлексов и ограничение функциональности коленных суставов. С первых дней жизни ребенок страдает от нарушения дыхания.
Спинально-мышечную атрофию важно дифференцировать с перинатальной энцефалопатией и родовыми травмами, но если при них состояние детей постепенно улучшается, то при СМА оно не меняется. Более того часто присоединяются осложнения, которые практически всегда приводят к смерти младенцев в течение первого месяца жизни.

Этот тип течения спинальной мышечной атрофии характеризуется очень тяжелым протеканием. Обычно она обнаруживается до 6-ти месяцев и сопровождается слабостью мышц, периодическими спазмами, что сложно заметить в связи с особенностями анатомии детей первого года жизни (присутствия ярко-выраженной подкожно-жировой клетчатки).
Также заболевание проявляется регулярно пробегающей по языку дрожью, снижением рвотного, сосательного, глотательного рефлексов. Это приводит к возникновению серьезных трудностей при кормлении. Присутствует нарушение слюноотделения, кашель. Ребенок часто громко кричит.
Эта форма спинально-мышечной атрофии может сопровождаться олигофренией и врожденными пороками сердца. Дети подвержены тяжелым нарушениям дыхания, развитию воспаления легких. В связи с этим более половины детей не доживает до 2 лет и только 10% могут отметить свой 5-летний юбилей. Причиной смерти становятся пневмония, остановка сердца или дыхательная недостаточность.
Заболевание обнаруживается у детей от 6 месяцев до 1,5–2 лет. Поэтому такую форму СМА часто называют поздней младенческой. Для нее типично:
- слабость и дрожь в мышцах;
- тремор пальцев, языка;
- скованность движений, обусловленная ограничением подвижности конечностей;
- задержка развития;
- недобор веса.
Дети с таким диагнозом способны самостоятельно сидеть, играть, есть, но стоять и передвигаться нет. К сожалению, патология склонна прогрессировать, что приводит к постепенному ослаблению мышц груди и шеи, следствием чего становится невозможность удерживать голову прямо и часто она безвольно свисает. Затем пропадают сухожильные рефлексы, слабеет голос и отмечаются нарушения акта глотания.
Длительность жизни при таком диагнозе составляет около 10–12 лет. Но треть больных погибает в возрасте до 4-х лет.
Спинальную мышечную атрофию этого вида диагностируют обычно после 2 лет. Она так же проявляется слабостью мышц, но не в такой степени как при СМА 1 или даже СМА 2. Больные могут самостоятельно стоять, но только в течение короткого периода времени. В связи с атрофией мышц это дается им с трудом.
Несмотря на имеющееся заболевание, до 10–12 лет ребенок развивается нормально, что может ввести его родных в заблуждение и вызвать сомнения в правильности поставленного диагноза. Но, достигая этого временного рубежа, возникают первые признаки СМА. Ребенок начинает спотыкаться чаще обычного, падает и не может выполнять физическую работу или заниматься спортом, часто сталкивается с переломами. Постепенно бег, а затем и ходьба даются все сложнее из-за возникновения ограничения подвижности суставов. Впоследствии подросток теряет способность передвигаться без инвалидного кресла.
Прогрессирование патологии приводит к возникновению тяжелого сколиоза, что влечет за собой изменение формы грудной клетки и появление трудностей при дыхании. Именно в этом таится главная угроза болезни для жизни.
К этому типу заболевания относят несколько разных не влияющих на продолжительность жизни, но приводящих к инвалидизации амиотрофий:
- бульбоспинальную Кеннеди;
- дистальную Дюшена-Арана;
- перонеальную Вюльпиана.
Их объединяет то, что первые клинические признаки заболевания проявляются в период от 16 до 60 лет, чаще в 35–40 лет. Это сопровождается угасанием сухожильных рефлексов и заметными спазмами мышц. При атрофии Дюшена-Арана сильнее всего страдают кисти, а для болезни Вюльпиана характерно изменение формы лопаток на крыловидную.
Симптомы СМА
Различные разновидности заболевания объединяет череда общих проявлений, хотя каждая из них имеет и специфичные симптомы. К числу общих признаков принадлежат:
- нарастающая мышечная слабость и постепенная атрофия;
- при тех видах спинальной мышечной атрофии, что обнаруживаются после года или двух лет, наблюдается деградация имеющихся физических достижений, к примеру, способности бегать, ходить;
- тремор пальцев, языка;
- искривление позвоночника;
- частое сохранение нормального психического развития и умственных способностей.
Статистика показывает, что чаще СМА поражает мальчиков.
Диагностика
Предельно информативным методом диагностики СМА считается генетический анализ. Его можно проводить ребенку и взрослому в любом возрасте, а с целью ранней диагностики его выполнение возможно еще на этапе внутриутробного развития. При невозможности проведения анализа ДНК и для окончательного подтверждения диагноза назначаются:
- биохимический анализ крови;
- гистологический анализ мышечных волокон;
- МРТ;
- электромиография;
- микроскопия спинного мозга;
- тандемная масс-спектрометрия.

Лечение спинально-мышечная атрофия
Больным назначается комплексная консервативная терапия, направленная на улучшение способности нервных импульсов проходить к мышцам и работы головного мозга. В этих целях рекомендуется прием:
- ноотропов;
- препаратов α-липоевой кислоты, ацетил-L-карнитина, α-глицерофосфохолина;
- витаминных комплексов, включающих, прежде всего, витамины группы В;
- средств, улучшающих обмен веществ.

Сегодня в разработке находятся специфические лекарственные средства, способные воздействовать на причину развития СМА – дефицит ряда белков. Но в данный момент они находятся на стадии испытаний. Пока что единственным способом хотя бы частично обеспечить организм необходимыми белками является соблюдение специальной диеты. Она подразумевает употребление продуктов, богатых на аминокислоты, а именно зерновых культур, орехов, кисломолочных продуктов, рыбы, мяса. Весьма полезно включение в меню шпината, брокколи, грейпфрутов. Особенно ценны блюда из бурого риса и овса.
Для поддержания мышечного тонуса рекомендованы:
- занятия ЛФК;
- массаж;
- физиотерапевтическое лечение;
- нейромышечная стимуляция.
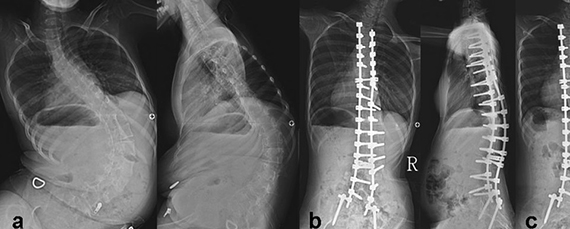
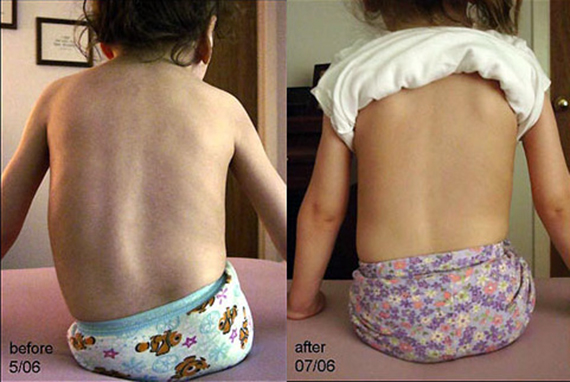
Современная медицина способна помочь пациентам с СМА за счет выравнивания позвоночника. Вы можете существенно повысить качество жизни и избавиться от болей с помощью хирургического лечения нейромышечного сколиоза. Наши спинальные хирурги способны грамотно провести операцию с учетом всех особенностей пациента и добиться предельно высоких результатов. Цены наших услуг приведены в прайсе.
Суть хирургического лечения нейромышечного сколиоза заключается в выполнении многоуровневой фиксации позвоночника с помощью специальных конструкций. Это предполагает изменение и закрепление в максимально приближенном к нормальному положению каждого сегмента искривленной части позвоночного столба.
Многоуровневая фиксация реализуется за счет установки многочисленных опорных элементов и выбора в качестве опорных точек крестца и таза и позвонков верхнегрудного отдела. Но часто ее проведение требуется практически по всей длине позвоночника, так как у больных спинальной мышечной атрофией сколиотические деформации достигают предельно тяжелых форм.
Она позволяет не только практически полностью выровнять позвоночник, но и равномерно распределить нагрузку на него, а также надежно удерживать его в новом положении. Благодаря этому больной избавляется от выраженного комфорта во время сидения и лежания, решаются психологические проблемы, спровоцированные выраженной деформацией позвоночного столба. Но главное достоинство операции заключается в устранении негативного влияния сколиоза на легкие и другие внутренние органы.
Стоимость коррекции сколиоза при СМА от 640 000 руб и зависит от:
— Тяжести заболевания (сколько времени пациент проведен в стационаре после операции)
— Фирмы производителя имплантов;
— Клиники (где будет проведена операция) и класса палаты.
Цена включает в себя:
— Прибывание в клинике до и после операции;
— Импланты.
— Операцию;
— Наркоз;
— Нейрофизиологический мониторинг
— Наблюдение и консультация на период реабилитации.
Все услуги клиники и стоимость приведены в прайсе.
На первичной консультации ответим на все интересующие вас вопросы, точно определим возможные риски и потенциальную пользу хирургического лечения и подарим вашему ребенку если не возможность ходить, то уверенно сидеть без болей и психологического дискомфорта.
Читайте также:
