
Спинальная амиотрофия верднига-гофмана после прививки

Амиатрофия Верднига-Гоффмана относится к орфанным (редким) патологиям, частота встречаемости которой 1:6000. Характеризируется крайне тяжелым течением и быстрым прогрессированием, приводящим к стойким и выраженным деформациям опорно-двигательного аппарата и летальному исходу.
- Что это?
- Причины заболевания
- I тип амиотрофии
- II тип
- III тип
- IV тип
- Диагностика
- Методы лечения
- Прогноз: сколько живут пациенты?
- Что нужно запомнить?
Что это?
Спинальная амиотрофия – генетическое заболевание, при котором обнаруживается мутации в 5 хромосоме в генах SMN1 и SMN2. Эти гены отвечают за сохранение и нормальное функционирование мотонейронов – нервов, которые отправляют импульс к скелетной мускулатуре. В результате отсутствия стимуляции мышц они атрофируются. У больных отмечается дегенеративные процессы в спинном мозге: разрушаются двигательные нейроны, нарушается функционирование передних рогов спинного мозга. В головном мозге изменения происходят в двигательных ядрах.
При мышечной амиотрофии Верднига мутирует ген SMN1, который препятствует гибели мотонейронов. Ген SMN2 выполняет эту функцию только частично и его возможности быстро истощаются. Течение и тяжесть патологии зависит от локализации и объема поражения в генном аппарате.

Фото 2
Спинальная амиотрофия вне зависимости от ее типа и формы – генетическое врожденное заболевание. Зачастую, первые признаки патологии обнаруживаются в младенческом возрасте либо еще во время беременности матери ребенка.
Одинаково распространена как у мужчин, так и женщин. Основное условие ее развития – наличие дефектного гена у обоих родителей, которые могут быть здоровыми и являются лишь носителями дефектной хромосомы.
В результате нарушения проводимости нервных импульсов от мотонейронов к мускулатуре происходит атрофия мышц нижних и верхних конечностей, диафрагмы, органов ЖКТ, сердца и другие. На фоне этого деформируются кости и суставы. Наиболее опасным проявлением является нарушения дыхательной функции и работы сердца.
В зависимости от тяжести течения, локализации дефектов и клинических проявлений выделяют 4 формы амиотрофии. Пациенты с любой формой амиотрофии являются инвалидами. Они не способны самостоятельно себя обслуживать, и нуждаются в постоянном уходе и медицинском наблюдении.
Причины заболевания
Основная причина развития спинальной амиотрофии – генетическая мутация у родителей и передача рецессивного гена плоду. Но, что привело к ее возникновению, ученым еще не известно. Мутация может произойти спонтанно, без видимых на то причин. Предполагаемыми причинами болезни являются следующие факторы:
- вирусные заболевания;
- нарушение экологии;
- прием во время беременности наркотиков, алкоголя, курение;
- воздействие терратогенных факторов;
- ионизирующее излучение.
В большинстве случаев родители узнают о том, что являются носителями опасного гена, только после рождения ребенка с диагнозом.
При некоторых вариантах амиотрофии толчком для прогрессирования мышечной дистрофии могут быть перенесенные вирусные заболевания, инсоляция, гормональный сбой.
I тип амиотрофии
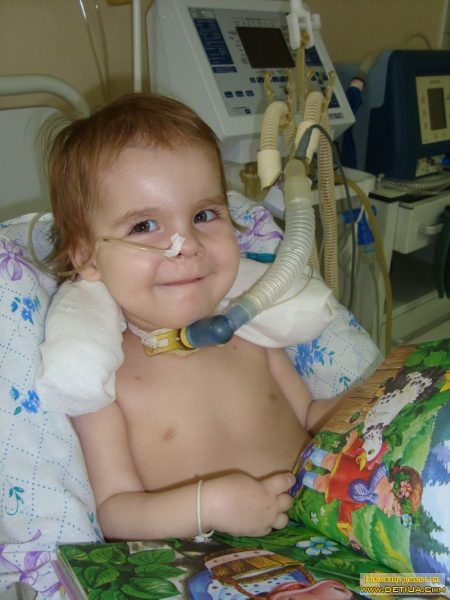
Наиболее злокачественный и распространенный тип патологии – амиотрофия Верднига-Гоффмана. Диагностируется у детей раннего возраста либо внутриутробно. У детей после рождения отмечается снижение и угасание всех рефлексов. Им сложно сосать грудь, из-за чего часто возникает необходимость кормить ребенка через зонд.
Мышечная гипотония приводит к слабости шейных мышц – дети не могут научиться держать головку, самостоятельно переворачиваться, сидеть, стоять и ходить. У большинства детей возникают трудности с глотанием. В большинстве случаев амиотрофию диагностируют в течение 6 месяцев после рождения. Родители обращают внимание на вялость ребенка и малоподвижность, прогрессирующее снижение мышечного тонуса. Также младенцы плохо набирают вес.
Характеризируется поражением диафрагмы, которая участвует в дыхательном акте. Многие пациенты самостоятельно не дышат. Из-за выраженной дыхательной недостаточности многие дети не доживают до года. Продлить им жизнь удается с помощью ИВЛ и питания через зонд. 95% детей умирают до 4 лет.
Патология сопровождается прогрессирующими деформациями опорно-двигательной системы.
Помимо поражения мышц и костей, часто выявляется отставание в умственном развитии.
Справка. Заподозрить диагноз можно еще во время беременности матери, когда отмечается слабое шевеление плода, нарушение сердечной деятельности, отставание в развитии.
II тип
Промежуточная амиотрофия. Диагностируется у детей 3 месяцев—1,5 года. В более раннем возрасте диагноз трудно установить из-за особенностей мышечной системы у детей. Из-за выраженной мышечной гипотонии и снижения сухожильных рефлексов младенцы не могут научиться ползать и ходить. Лишь в 25% возможно, что ребенок сможет самостоятельно сесть.
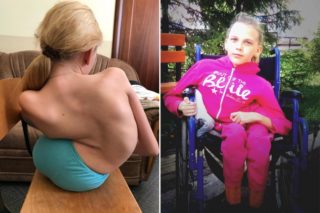
Сопровождается поражением костно-суставной системы. У многих искривляется позвоночник, деформируется грудная клетка, атрофируются суставы рук и ног. Родители отмечают похудение ребенка либо прекращение набора веса.
Особенность этого типа амиотрофии – возможное внезапное прекращение ее развития. Несмотря на ремиссию, утратившиеся способности практически невозможно восстановить. Прогрессирование болезни может возобновиться либо ускориться после перенесенного ОРВИ или ОРЗ и других заболеваний, снижающих иммунитет. Из-за атрофии диафрагмы нарушается работа легких, ослабевает работа легких, из-за чего возникает одышка и тахикардия. Вовлечение в процесс мускулатуры органов пищеварения, бульбарные нарушения приводят к неспособности самостоятельно принимать пищу.
III тип
Болезнь Кугельберга-Веландера – вариант поздней амиотрофии. При данной форме диагноза первые признаки возникают после двух лет, либо во взрослом возрасте. Дети, которые уже умеют ходить, внезапно становятся неловкими, жалуются на боль в ногах при хождении. Они слишком часто падают, неуверенно ходят и практически перестают бегать. Многие родители замечают изменения походки и внезапно появившуюся неуклюжесть. Вначале развития патологии поражаются мышцы ног, затем атрофируются мышцы верхних конечностей и других частей тела. Из-за снижения объема мышц также отмечается снижение веса.
У некоторых больных может наступить период ремиссии, в который прекращается прогрессирование диагноза. Этот период может продлиться от нескольких недель, до нескольких десятков лет.
Нарушение дыхательной функции наиболее опасное проявление атрофии мускулатуры.
Со стороны психики и умственного развития нарушения не выявляются.
IV тип
Первые признаки и симптомы появляются в 30-50 лет. Основными жалобами является слабость мышц, тремор. Из-за атрофии появляются контрактуры в области суставов, с ограничением их подвижности. Больные резко худеют.
Часто сопровождается искривлением позвоночника и деформацией грудной клетки.
Первыми в процесс вовлекаются мышцы ног, после постепенно вовлекаются руки. Как правило, дыхательная и глотательная функция не нарушена.
Большинство пациентов самостоятельно ходят, но хромают, испытывают боль и дискомфорт. В редких случаях прогрессирование амиотрофии приводит к потере способности ходить, и больные вынуждены передвигаться на инвалидной коляске.
Диагностика
Основным методом диагностики является генетическое исследование. Для проведения тестирования используют кровь. В некоторых случаях для подтверждения диагноза проводят биопсию пораженных тканей.
Для определения тяжести болезни, объема поражений и нарушения функционирования мышечной системы проводят электромиографию.
Также обследовать необходимо и других близких родственников пациента.
Важно! Для пренатальной диагностики исследуют околоплодные воды. Такой метод применяется только при наличии симптомов и генетической предрасположенности у родителей.
Методы лечения
Генетическое заболевание является неизлечимым и практически не поддается коррекции. При прогрессирующем течении практически невозможно предотвратить и остановить мышечную атрофию. Несмотря на развитие генной инженерии, ученым не удалось создать лекарство, способное устранить генетический сбой.
Лечение направлено на поддерживание жизненно важных функций организма, облегчение состояния больного и предотвращение деформации скелета. Для этого применяют следующие методы:
- искусственная вентиляция легких – показана при дыхательной недостаточности;
- физиопроцедуры – массаж, ЛФК, электрофорез, лечебные ванны и другие;
- ношение корсета и других ортопедических приспособлений;
- витаминотерапия;
- применение ноотропов и препаратов, улучшающих питание нервной ткани: актовегин, трентал, церебролизин и другие.
Паллиативная терапия направлена на облегчение состояние тяжелобольных пациентов.
Ученые всего мира пытаются создать препарат для лечения патологии. Известно, что уже разработано несколько препаратов, которые возможно сумеют помочь пациентам. Они находятся на стадии клинических испытаний и их эффективность и безопасность для организма только изучается.
Родителям детей с тяжелым, неподдающимся лечению, диагнозом, для рождения здорового ребенка необходимо планировать беременность с репродуктологом. Искусственное оплодотворение, использование донорской спермы или яйцеклетки, экстракорпоральное оплодотворение – одни из способов родить здорового ребенка носителям дефектного гена.
Прогноз: сколько живут пациенты?
Спинальная амиотрофия крайне тяжелое заболевание, которое существенно ухудшает качество жизни больных и является одной из причин младенческой смерти. Пациенты с таким диагнозом являются инвалидами и зачастую не в силах самостоятельно себя обслуживать.
Многие пациенты не могут ходить и стоять, передвигаясь с помощью инвалидной коляски. При поражении рук больные не способны делать элементарных вещей: есть, держать небольшие предметы, читать, умываться и прочее. Они нуждаются в дополнительном уходе и постоянном медицинском наблюдении. При тяжелом течении они дышать лишь с помощью аппарата ИВЛ и питаются через зонд.
Продолжительность жизни во многом зависит от формы заболевания. Наиболее тяжелой является амиотрофия Верднига, при которой больные дети редко достигают 4 лет. Пациенты со вторым типом болезни редко доживают до совершеннолетия. Наиболее благополучным являются второй и третий тип болезни, при которых пациенты достигают зрелого возраста.
В настоящее время спинальная амиотрофия относится к неизлечимым заболеваниям, часто приводит к летальному исходу.
Родители детей со СМА сначала живут с абсолютно здоровыми детьми
Именно в этот период родители замечают, что с ребенком что-то не то. Ведь большинство детей со СМА выписывают из роддома абсолютно здоровыми, так было и с моей дочкой. Сначала все прекрасно – и общие показатели, и прибавка в весе. Мы все, родители детей со СМА, какое-то время живем с абсолютно здоровыми детьми – так думаем не только мы, но и наблюдающие врачи, пока не появятся первые признаки, указывающие на болезнь ребенка. У каждого в разное время.
Кто виноват, что наш ребенок болен?
Но на самом деле, ребенок, несущий генетическую мутацию, уже болен. И когда становится понятно, что ребенок не набирает навыки, которые уже должны быть в его возрасте, или набирает, но не совсем так, как положено (есть разные типы СМА, проявляющиеся по-разному), родители начинают искать причину.

Ольга Германенко. Фото: Фейсбук
Говорить о точной взаимосвязи прививок и старта СМА нельзя
Среди родителей детей со СМА очень многие считают, что именно прививка дала старт болезни. Они думают, что не сделай мы эту прививку, ребенок смог бы набрать больше навыков и старт болезни был бы позже. На данный момент невозможно ни согласиться, ни отрицать взаимосвязь проявлений первых симптомов СМА (которые в любом случае бы проявились) – таких исследований просто не проводилось. И мы не знаем, так это или нет.
Говорить о точной взаимосвязи прививок и старта СМА нельзя, более того – ни в Европе, ни в США спинальная мышечная атрофия не является противопоказанием к проведению прививок. Эти дети прививаются по графику точно так же, как обычные здоровые дети, более того, график вакцинации для них даже более обширен и включает несколько дополнительных вакцин.
Лично я не сторонник и не противник вакцинации. На вопрос стоит или нет делать прививки, у меня нет готового ответа. Но все родители при любой вакцинации должны смотреть не на диагноз – СМА это, или ДЦП, или любая другая патология, а на состояние конкретного ребенка непосредственно перед прививкой.
Необходимо выполнить все необходимые обследования и анализы, оценить факторы окружения ребенка, семейную историю. Если есть малейшее сомнение относительно состояния здоровья ребенка в момент перед вакцинацией, прививку стоит отложить на момент, когда она будет безопасной.
К сожалению, сейчас в России сложилась дурная практика – обычно никто внимательно не смотрит ребенка перед прививкой, ведь есть график, в который нужно уложиться. Есть план и практически отсутствует индивидуальный подход к ведению пациентов.
Возможно, в том числе, и этот массовый подход к проблемам вакцинации приводит нас к тому, что многие родители считают, что именно прививка стала стимулирующим фактором в развитии болезни. Наша медицинская система, к сожалению, не чувствительна к особенным проявлениям индивидуального пациента, это большая проблема нашей страны – и социальной, и медицинской сферы.
Юлия Самойлова: СМА или последствия прививки
Мне сложно говорить о ситуации, которой я не знаю из первых рук, т.к. контакта с самой Юлей у меня не было. Здесь я могу рассуждать только об общих, хорошо известных вещах касательно СМА. Как развивалась болезнь именно у Юлии, у меня нет данных, но в публикациях она рассказывает, что генетический диагноз СМА был установлен в 13 лет, а первые признаки заболевания у нее появились примерно в год.
То есть в это время начались клинические проявления, симптомы болезни. Юле 27 лет, и речь идет о начале 90-х годов. В те годы диагноз СМА генетически поставить было невозможно. Вспомним, что до 1995 года генетические тестирования не проводились нигде в мире, так как в то время еще не был известен ген SMN, отвечающий за болезнь СМА. В нашей стране анализ стал доступен уже в 2000-х.
Спинальная мышечная атрофия (СМА) – генетическое заболевание, поражающее область нервной системы, отвечающей за контроль движений произвольных (скелетных) мышц.
Спинальная мышечная атрофия у детей впервые была описана Вердниг ом в 1891 году . Вердниг представил описание патоморфологических изменений различных групп мышц, периферических нервов и спинного мозга, отметив симметричную атрофию клеток передних рогов спинного мозга и передних корешков. В 1892 г. Хоффман обосновал нозологическую самостоятельность заболевания. В дальнейшем Вердниг и Хоффман ( 1893 ) доказали, что заболевание сопровождается дегенерацией клеток передних рогов спинного мозга.
Еще один вопрос, который поднимается в связи с диагнозом Юлии – это форма болезни. С пинальная мышечная атрофия Верднига-Гоффмана, как пишут СМИ, и это не совсем верно. СМА Верднига-Гоффмана или СМА 1 типа, – это самый тяжелый тип заболевания, обычно проявляется раньше, и дети с этой формой не могут самостоятельно сидеть без поддержки, а средняя продолжительность жизни без применения ИВЛ – около двух лет.
Это явно не тот тип СМА, который у Юлии. Просто раньше в нашей российской медицине любой тип спинальной амиотрофии называли болезнью Верднига-Гоффмана, и только буквально последние 10 лет ситуация изменилась и стала соответствовать общемировой практике. Все наши пациенты, кому уже больше 18 лет, имели запись в медкарте, что у них болезнь Верднига-Гоффмана, хотя на самом деле их болезнь это II или III тип, в зависимости от клинических проявлений болезни.

Против СМА у нас не было ни шанса, теперь есть лечение
Участие Юлии Самойловой в Евровидении для довольно обширного сообщества СМА-пациентов в России стало прекрасной новостью. Ведь это шанс не только в очередной раз почувствовать, что жизнь болезнью не ограничивается, она гораздо шире и наполненнее, и никакая болезнь не должна помешать исполнению мечты, но и привлечь внимание к нуждам и потребностям пациентов с этим заболеванием.
К сожалению, большинство больных со СМА в настоящее время лишены возможности получить необходимые им помощь и поддержку, как социальную, так и медицинскую. Нужное для поддержания здоровья и жизни наших подопечных дорогостоящее медицинское оборудование не входит в государственные программы помощи, и его приходится закупать за свой счет или за счет благотворителей. А стоимость, например, откашливателя или аппарата вентиляции легких начинается от 300 тысяч рублей.
Предоставляемые государством технические средства реабилитации не отвечают индивидуальным потребностям пациентов со СМА, нуждающихся в индивидуальном подборе в соответствии с их состоянием. Проблем много и очень бы хотелось, чтобы постепенно они все же решались не только за счет благотворительности, но и социальной ответственностью государства перед своими гражданами.
Но есть и еще причина, почему сейчас особенно для нас важно распространение информации о ситуации с людьми, больными СМА, – это появление новой перспективной терапии. В конце прошлого года, 24 декабря, FDA (Управление по санитарному надзору за качеством пищевых продуктов и медикаментов, США) одобрило первое лекарство для лечения СМА.
И несмотря на то, что это лекарство окончательно болезнь не излечивает, оно является поддерживающей терапией, способной значительно облегчить состояние больных СМА, приостановить прогрессирование заболевания. А у некоторых детей, участников клинических испытаний препарата, были отмечены даже значительные улучшения моторных функций.
Это новая терапия, сейчас она проходит одобрение в Европейском медицинском агентстве, аналогичном американскому, и пока это лечение доступно только в США (и пока далеко не всем детям со СМА, сейчас идут процессы согласования оплаты лечения страховыми компаниями), потому что по озвученным ценам это лекарство одно из самых дорогих в мире, значительно дороже других препаратов для орфанных заболеваний, известных своими высокими ценами.
Первое лекарство от спинальной мышечной атрофии (СМА) будет стоить 125 000 $ за одну инъекцию. В первый год лечения пациент должен получить шесть таких инъекций (это 750 000 $), а затем ежегодно тратить на лечение 375 000 $.
Тем не менее, это единственная одобренная терапия СМА, и это прорыв. Заболевание, в борьбе с которым ранее у нас не было ни шанса, теперь имеет свое лечение. Мы очень надеемся, что препарат будет зарегистрирован и в нашей стране, и наши больные СМА тоже смогут его получать. А пока мы наблюдаем за успехами терапии у американских детей, которые либо участвовали в клинических испытаниях, либо применяют препарат сейчас.
Не надо забывать о самой Юлии и ее силе духа
Сейчас в прессе появляется масса публикаций, связанных с участием Юлии Самойловой в конкурсе, но, к большому сожалению, все они в основном посвящены совсем другим вопросам, чем должны были бы. Общество активно обсуждает проблему вакцинации, физическое состояние Юлии и факт ее болезни, еще охотнее – возможные политические составляющие, сопряженные с ее участием в конкурсе.
Но на что мы действительно должны обратить внимание — это на саму Юлию – на ее творчество, на ее жизнь, полную побед и, надеюсь, радостей. На силу духа молодой россиянки, которая вопреки болезни и несмотря на все сложности, с которыми сталкиваются люди с инвалидностью в нашей стране, живет полноценной, насыщенной жизнью. Вот что действительно стоит внимания и достойно восхищения.
Мы должны стремиться к тому, чтобы люди на колясках могли ощущать себя в нашем обществе, на улице и в публичном пространстве не инвалидами, не кем-то особенным, а просто людьми. Обычными людьми со своими интересами, хобби, мечтами.
Хотелось бы еще подчеркнуть, что сам факт, что Юлия Самойлова едет на Евровидение, очень значим, и неважно, что там – СМА это, или осложнение от прививки, или ее просто используют в политических маневрах. Просто не надо забывать о самой Юлии. Ведь несмотря на такую сложную ситуацию, она очень многого добилась.
С такой болезнью очень сложно справляться, а тот факт, что сама Юлия не ищет контактов с такими же больными, а справляется сама, говорит о ее самодостаточности и сильной воле. Это тот случай, когда факт самой инвалидности не ставится на первое место, а значение имеют именно возможности человека. Не ограниченные возможности, не инвалидная коляска, не СМА, а именно достижения и успехи Юлии в музыке имеют значение.
И я надеюсь, что мы все будем видеть в Юлии именно талантливую певицу и слушать и погружаться в ее музыку, а не видеть за Юлей диагноз. Потому что это просто нечестно по отношению к ней.
Давайте все просто пожелаем Юлии успеха и будем болеть за нее на конкурсе!

Работа фонда имеет два основных направления: оказание помощи непосредственно самим больным СМА и их близким и работа на системные изменения с ситуацией оказания помощи и поддержки семьям, в которых воспитываются дети со СМА и взрослым пациентам.
Помочь можно, оформив разовое или регулярное пожертвование на специальной странице фонда или отправив СМС на короткий номер 3443 со словом СМА и через пробел указать сумму пожертвования цифрами, например, СМА 300.

Болезнь Верднига-Гоффмана
Спинальная амиотрофия I, II, III типа (болезнь Верднига-Гоффмана, болезнь Кюгельберга-Веландер, САМ, SMA)
Проксимальная спинальная амиотрофия I, II III типа (САМ I-III) - одно из наиболее частых наследственных заболеваний с аутосомно-рецессивным типом наследования, с частотой встречаемости 1 на 6000-10000 новорожденных. Основной механизм развития клинических признаков связан с прогрессирующей дегенерацией мотонейронов передних рогов спинного мозга, что выражается в атрофии проксимальных мышц конечностей, в первую очередь.
Выделяют три формы заболевания на основе возраста начала, тяжести течения и продолжительности жизни.
СAМ I (болезнь Верднига-Гоффмана, OMIM*253300) - наиболее тяжелая форма, первые симптомы можно нередко выявить еще во внутриутробном периоде по слабому шевелению плода. У значительного числа больных этой формой отчетливые клинические проявления отмечаются до 6-ти месячного возраста и характеризуются выраженными признаками вялого паралича мышц конечностей и туловища, с вовлечением в процесс дыхательной мускулатуры. Дети не держат голову, не сидят самостоятельно.
СAМ II (промежуточная форма, OMIM*253550) имеет более позднее начало, как правило, после 6 месяцев. Больные дети могут сидеть, но никогда не достигают способности ходить самостоятельно. Прогноз в этих случаях зависит от степени вовлечения в патологический процесс респираторных мышц.
При СAМ III (болезнь Кюгельберга-Веландер, OMIM*253400) первые симптомы у пациентов появляются после 18 месяцев, они могут стоять и ходить самостоятельно.
Ген, ответственный за возникновение САМ I-III, названный SMN (survival motor neuron gene), расположен в районе 5q13 и представлен двумя высокогомологичными копиями (теломерной - SMN1 и центромерной - SMN2). У 96% пациентов с различными типами САМ I-III регистрируется делеция гена SMN1.
В Центре Молекулярной Генетики проводится прямая и косвенная ДНК-диагностика САМ I-III. Прямая диагностика основана на амплификации фрагментов 7 и 8 экзонов обоих генов с последующей рестрикцией, дающей возможность зарегистрировать наличие/отсутствие соответствующих экзонов генов SMN1 и SMN2, а также определить их соотношение.
Таким образом, прямая ДНК-диагностика помимо подтверждения диагноза САМ I-III с помощью молекулярно-генетических методов, дает возможность зарегистрировать гетерозиготное носительство мутации, что имеет большое значение для семей, где материал больного ребенка недоступен, а также для здоровых сибсов пациентов с диагнозом САМ I-III для их дальнейшего медико-генетического консультирования. Учитывая высокую частоту носительства заболевания (приблизительно 1 на 40 человек) целесообразно выявление гетерозиготного носительства делеции гена SMN1 в популяции. Кроме того, возможно проведение косвенной ДНК-диагностики САМ I-III с использованием полиморфных ДНК-маркеров. Проведение дородовой ДНК-диагностики САМ I-III прямыми и косвенными методами одновременно снижает риск рождения больного ребенка практически до 0% .
Клиническая картина
Современная медицина выделяет три типа заболевания. Это врожденная форма, ранняя детская и поздняя, отличающаяся периодом проявления первых клинических симптомов и скоростью протекания миодистрофического процесса.
Болезнь злокачественная, быстро прогрессирующая. Смерть может наступить до 9-летнего возраста. Одна из основных причин смерти – это тяжелые соматические расстройства, проявляющиеся всердечно-сосудистой и дыхательной недостаточности, которые обусловлены слабостью и снижением участия мускулатуры грудной клетки в физиологии дыхания.
Первые признаки болезни в случае ранней детской формы обычно проявляются во втором полугодии жизни. В течение первых месяцев моторное развитие удовлетворительное. Ребенок во время начинает держать голову, садится, иногда сам стоит. Развитие заболевания проходит подостро, обычно после пищевой интоксикации или перенесенной инфекции. Первоначально вялые парезы наблюдаются в ногах, затем они быстро переходят на мышцы руки и туловища. Проявляются диффузные мышечные атрофии, сочетающиеся с мелким тремором пальцев, сухожильными контрактурами и фасцикуляциями языка. Глубокие рефлексы и тонус мышц снижается. На более поздних стадиях развиваются явления бульбарного паралича и генерализованная мышечная гипотония.
Болезнь имеет злокачественный характер, хотя протекает мягче, нежели врожденная форма. Как правило, смерть наступает в 14-15 летнем возрасте.
Спинальная мышечная атрофия 1 типа – это тяжелое заболевание, при котором шансы на излечение отсутствуют. В настоящее время самым эффективным способом борьбы с патологией остается дородовая диагностика и аборт по медицинским показаниям. Но если предотвратить рождение больного ребенка не удалось или для родителей это неприемлемо, то семье придется столкнуться с трудностями ухода и лечения ребенка-инвалида.
Этиология амиотрофии Верднига-Гоффмана
При генетических нарушениях у пациентов постепенно отмирают двигательные нейроны спинного мозга. Без достаточной иннервации мышцы конечностей и внутренних органов не могут выполнять свою функцию и нормально питаться. В них тоже начинаются процессы отмирания. Это и есть спинальная амиотрофия.
Спровоцировать генетические поломки могут различные вредные факторы – воздействие токсических веществ и радиации на половые клетки, неблагоприятные факторы и заболевания матери во время беременности, в меньшей степени – курение и алкоголизм родителей.
Чаще всего это мутация в первом поколении, поскольку пациенты с симптомами спинальной амиотрофии Верднига-Гоффмана крайне редко доживают до репродуктивного возраста.
Симптомы заболевания

Патология проявляется очень рано – первые признаки становятся видны до того, как малышу исполнится полгода. В особенно тяжелых случаях симптомы отмечаются еще у плода в виде слабых или очень слабых шевелений. При рождении у ребенка может отмечаться сниженный тонус мышц. Чаще всего болезнь диагностируется в 2-3 месяца, когда малыш не начинает вовремя держать голову.
Характерные особенности больных:
- прогрессирующая мышечная слабость;
- прекращение активных движений вплоть до паралича;
- сниженный сосательный рефлекс, постоянное недоедание;
- сохранение чувствительности кожи;
- поражение больших групп мышц, постепенное расширение пораженной области.
Спинальная амиотрофия 1 типа отличается стремительным развитием. В течение нескольких месяцев присоединяются тяжелые осложнения – дыхательная недостаточность и аспирационная пневмония. Из-за сниженного сосательного и глотательного рефлекса малыши не могут полноценно питаться, единственный способ их накормить – внутривенный.
Этому состоянию обычно сопутствует целый букет других врожденных патологий – деформации костей, нарушения формирования суставов и кровеносных сосудов, гидроцефалия, олигофрения. Существуют и менее злокачественные формы болезни – они прогрессируют медленнее, имеют меньше сопутствующих патологий, первые проявления возникают позже. При 2 типе пациенты могут дожить до подросткового возраста, при 3 – до зрелости.
Как ставится диагноз
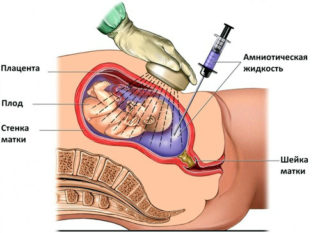
Выявить спинальную мышечную атрофию Верднига-Гоффмана можно до или после рождения ребенка. Пренатальная диагностика основана на выявлении генетического дефекта. Для этого требуется получить генетический материал ребенка из пуповинной крови, амниотической жидкости или ворсин хориона. Инвазивные методы диагностики опасны для развития беременности, поэтому родители часто отказываются от них. Если патология выявлена, это показание к аборту.
Рождение детей со спинальной мышечной атрофией (СМА) Верднига-Гоффмана возможно по нескольким причинам. Не во всех больницах есть возможность дородовой диагностики болезни и генетического консультирования родителей до зачатия малыша. Родители проводят анализ в основном по собственному желанию и могут до момента родов не знать о том, что малыш будет тяжело болен. Иногда диагноз ставится на позднем сроке, когда прерывать беременность поздно. Играет роль и принципиальное неприятие абортов у некоторых родителей – даже зная о будущей патологии, они принимают решение рожать.
После родов диагноз ставит неонатолог или детский невролог. Важен неврологический статус маленького пациента – нарушение двигательной активности и угасание рефлексов при сохранении чувствительности. Для постановки окончательного диагноза назначается анализ ДНК. МРТ и КТ позвоночника важны, чтобы отличить СМА от других патологий. Нарушения в двигательных ядрах спинного мозга не визуализируются.
Лечение мышечной атрофии и профилактика
Лечение спинальной амиотрофии Верднига-Гоффмана симптоматическое. Избежать развития болезни или существенно замедлить нарастающие нарушения невозможно, но можно снять выраженность симптомов. Терапия проводится тремя основными способами – медикаментозное лечение, лечебная физкультура и устройства, облегчающие существование больных детей и их родственников.
Из медикаментозных средств при спинальной мышечной атрофии 1 типа применяются ноотропные препараты и нейрометаболиты – они улучшают передачу импульсов в нервно-мышечных синапсах. При нарушениях глотательного рефлекса единственный возможный путь введения препаратов – инъекционный.
Лечебная физкультура и массаж помогают повысить тонус мышц, улучшить кровоснабжение и замедлить процессы атрофии. Быстро прогрессирующая спинальная амиотрофия при болезни Верднига-Гоффмана не дает детям возможности выполнять активные движения. Гимнастика построена только на пассивных действиях – их осуществляют родители.
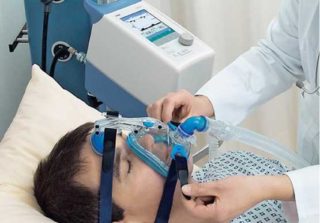
На поздних этапах заболевания жизнь пациентов значительно облегчают портативные аппараты ИВЛ. Это позволяет детям находиться дома в привычной обстановке. У пациентов с более легкими формами, проявляющимися после полугода или в подростковом возрасте, применяются автоматизированные инвалидные коляски.
Поскольку причина спинальной амиотрофии в генетическом дефекте, предотвратить заболевание можно только одним путем – не допускать рождения детей, у которых диагностирована патология. Важную роль играет генетическое консультирование. Пациенты с самой легкой формой генетически обусловленной гибели нейронов (3 тип, болезнь Кугельберга-Валандера) доживают до репродуктивного возраста и имеют шанс рождения детей с более тяжелыми формами, чем у них самих.
Общая проблема всех заболеваний, которые можно диагностировать до родов – не для всех родителей приемлемо прерывание беременности даже по медицинским показаниям. Кроме того, тест на спинальную мышечную атрофию 1 типа у детей не является обязательным. Большая часть диагнозов этого заболевания ставится уже после рождения ребенка, и родители оказываются перед печальным фактом, что ребенок страдает неизлечимым заболеванием.
Осложнения и прогноз
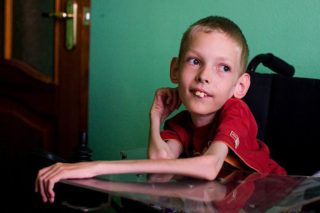
Болезнь Верднига-Гоффмана быстро прогрессирует – чем раньше оно проявилось, тем быстрее придет к летальному исходу. Маленькие пациенты неизбежно гибнут. Средний срок жизни таких детей – около двух лет, максимальная продолжительность – до 8 лет, если первые признаки болезни появились в 5-6 месяцев.
Другие виды спинальной амиотрофии, не относящиеся к болезни Верднига-Гоффмана, имеют более благоприятный прогноз. Сколько живут пациенты, зависит от возраста, в котором манифестировала патология. Раннее начало – неблагоприятный признак, если симптомы появились в подростковом возрасте (3 тип), то пациент может дожить до старости только с частичной утратой двигательной активности.
Основные осложнения спинномозговой атрофии Верднига-Гоффмана:
- нарушение сосательного рефлекса и общая гипотрофия ребенка;
- нарушение глотания, попадание пищевых масс в дыхательные пути и аспирационная пневмония;
- атрофия межреберных мышц и диафрагмы, дыхательная недостаточность и полная невозможность самостоятельного дыхания.
Последнее – основная причина смерти пациентов.
Нарушение функций спинного мозга крайне опасно для жизни и не дает никаких шансов на выживание. Единственная возможность предотвращения – совершенствование методов дородовой диагностики и генетического консультирования.
Читайте также:
