
Как наследуется спинальная амиотрофия

Амиатрофия Верднига-Гоффмана относится к орфанным (редким) патологиям, частота встречаемости которой 1:6000. Характеризируется крайне тяжелым течением и быстрым прогрессированием, приводящим к стойким и выраженным деформациям опорно-двигательного аппарата и летальному исходу.
- Что это?
- Причины заболевания
- I тип амиотрофии
- II тип
- III тип
- IV тип
- Диагностика
- Методы лечения
- Прогноз: сколько живут пациенты?
- Что нужно запомнить?
Что это?
Спинальная амиотрофия – генетическое заболевание, при котором обнаруживается мутации в 5 хромосоме в генах SMN1 и SMN2. Эти гены отвечают за сохранение и нормальное функционирование мотонейронов – нервов, которые отправляют импульс к скелетной мускулатуре. В результате отсутствия стимуляции мышц они атрофируются. У больных отмечается дегенеративные процессы в спинном мозге: разрушаются двигательные нейроны, нарушается функционирование передних рогов спинного мозга. В головном мозге изменения происходят в двигательных ядрах.
При мышечной амиотрофии Верднига мутирует ген SMN1, который препятствует гибели мотонейронов. Ген SMN2 выполняет эту функцию только частично и его возможности быстро истощаются. Течение и тяжесть патологии зависит от локализации и объема поражения в генном аппарате.

Фото 2
Спинальная амиотрофия вне зависимости от ее типа и формы – генетическое врожденное заболевание. Зачастую, первые признаки патологии обнаруживаются в младенческом возрасте либо еще во время беременности матери ребенка.
Одинаково распространена как у мужчин, так и женщин. Основное условие ее развития – наличие дефектного гена у обоих родителей, которые могут быть здоровыми и являются лишь носителями дефектной хромосомы.
В результате нарушения проводимости нервных импульсов от мотонейронов к мускулатуре происходит атрофия мышц нижних и верхних конечностей, диафрагмы, органов ЖКТ, сердца и другие. На фоне этого деформируются кости и суставы. Наиболее опасным проявлением является нарушения дыхательной функции и работы сердца.
В зависимости от тяжести течения, локализации дефектов и клинических проявлений выделяют 4 формы амиотрофии. Пациенты с любой формой амиотрофии являются инвалидами. Они не способны самостоятельно себя обслуживать, и нуждаются в постоянном уходе и медицинском наблюдении.
Причины заболевания
Основная причина развития спинальной амиотрофии – генетическая мутация у родителей и передача рецессивного гена плоду. Но, что привело к ее возникновению, ученым еще не известно. Мутация может произойти спонтанно, без видимых на то причин. Предполагаемыми причинами болезни являются следующие факторы:
- вирусные заболевания;
- нарушение экологии;
- прием во время беременности наркотиков, алкоголя, курение;
- воздействие терратогенных факторов;
- ионизирующее излучение.
В большинстве случаев родители узнают о том, что являются носителями опасного гена, только после рождения ребенка с диагнозом.
При некоторых вариантах амиотрофии толчком для прогрессирования мышечной дистрофии могут быть перенесенные вирусные заболевания, инсоляция, гормональный сбой.
I тип амиотрофии
Наиболее злокачественный и распространенный тип патологии – амиотрофия Верднига-Гоффмана. Диагностируется у детей раннего возраста либо внутриутробно. У детей после рождения отмечается снижение и угасание всех рефлексов. Им сложно сосать грудь, из-за чего часто возникает необходимость кормить ребенка через зонд.
Мышечная гипотония приводит к слабости шейных мышц – дети не могут научиться держать головку, самостоятельно переворачиваться, сидеть, стоять и ходить. У большинства детей возникают трудности с глотанием. В большинстве случаев амиотрофию диагностируют в течение 6 месяцев после рождения. Родители обращают внимание на вялость ребенка и малоподвижность, прогрессирующее снижение мышечного тонуса. Также младенцы плохо набирают вес.
Характеризируется поражением диафрагмы, которая участвует в дыхательном акте. Многие пациенты самостоятельно не дышат. Из-за выраженной дыхательной недостаточности многие дети не доживают до года. Продлить им жизнь удается с помощью ИВЛ и питания через зонд. 95% детей умирают до 4 лет.
Патология сопровождается прогрессирующими деформациями опорно-двигательной системы.
Помимо поражения мышц и костей, часто выявляется отставание в умственном развитии.
Справка. Заподозрить диагноз можно еще во время беременности матери, когда отмечается слабое шевеление плода, нарушение сердечной деятельности, отставание в развитии.
II тип
Промежуточная амиотрофия. Диагностируется у детей 3 месяцев—1,5 года. В более раннем возрасте диагноз трудно установить из-за особенностей мышечной системы у детей. Из-за выраженной мышечной гипотонии и снижения сухожильных рефлексов младенцы не могут научиться ползать и ходить. Лишь в 25% возможно, что ребенок сможет самостоятельно сесть.
Сопровождается поражением костно-суставной системы. У многих искривляется позвоночник, деформируется грудная клетка, атрофируются суставы рук и ног. Родители отмечают похудение ребенка либо прекращение набора веса.
Особенность этого типа амиотрофии – возможное внезапное прекращение ее развития. Несмотря на ремиссию, утратившиеся способности практически невозможно восстановить. Прогрессирование болезни может возобновиться либо ускориться после перенесенного ОРВИ или ОРЗ и других заболеваний, снижающих иммунитет. Из-за атрофии диафрагмы нарушается работа легких, ослабевает работа легких, из-за чего возникает одышка и тахикардия. Вовлечение в процесс мускулатуры органов пищеварения, бульбарные нарушения приводят к неспособности самостоятельно принимать пищу.
III тип
Болезнь Кугельберга-Веландера – вариант поздней амиотрофии. При данной форме диагноза первые признаки возникают после двух лет, либо во взрослом возрасте. Дети, которые уже умеют ходить, внезапно становятся неловкими, жалуются на боль в ногах при хождении. Они слишком часто падают, неуверенно ходят и практически перестают бегать. Многие родители замечают изменения походки и внезапно появившуюся неуклюжесть. Вначале развития патологии поражаются мышцы ног, затем атрофируются мышцы верхних конечностей и других частей тела. Из-за снижения объема мышц также отмечается снижение веса.
У некоторых больных может наступить период ремиссии, в который прекращается прогрессирование диагноза. Этот период может продлиться от нескольких недель, до нескольких десятков лет.
Нарушение дыхательной функции наиболее опасное проявление атрофии мускулатуры.
Со стороны психики и умственного развития нарушения не выявляются.
IV тип
Первые признаки и симптомы появляются в 30-50 лет. Основными жалобами является слабость мышц, тремор. Из-за атрофии появляются контрактуры в области суставов, с ограничением их подвижности. Больные резко худеют.
Часто сопровождается искривлением позвоночника и деформацией грудной клетки.
Первыми в процесс вовлекаются мышцы ног, после постепенно вовлекаются руки. Как правило, дыхательная и глотательная функция не нарушена.
Большинство пациентов самостоятельно ходят, но хромают, испытывают боль и дискомфорт. В редких случаях прогрессирование амиотрофии приводит к потере способности ходить, и больные вынуждены передвигаться на инвалидной коляске.
Диагностика
Основным методом диагностики является генетическое исследование. Для проведения тестирования используют кровь. В некоторых случаях для подтверждения диагноза проводят биопсию пораженных тканей.
Для определения тяжести болезни, объема поражений и нарушения функционирования мышечной системы проводят электромиографию.
Также обследовать необходимо и других близких родственников пациента.
Важно! Для пренатальной диагностики исследуют околоплодные воды. Такой метод применяется только при наличии симптомов и генетической предрасположенности у родителей.
Методы лечения
Генетическое заболевание является неизлечимым и практически не поддается коррекции. При прогрессирующем течении практически невозможно предотвратить и остановить мышечную атрофию. Несмотря на развитие генной инженерии, ученым не удалось создать лекарство, способное устранить генетический сбой.
Лечение направлено на поддерживание жизненно важных функций организма, облегчение состояния больного и предотвращение деформации скелета. Для этого применяют следующие методы:
- искусственная вентиляция легких – показана при дыхательной недостаточности;
- физиопроцедуры – массаж, ЛФК, электрофорез, лечебные ванны и другие;
- ношение корсета и других ортопедических приспособлений;
- витаминотерапия;
- применение ноотропов и препаратов, улучшающих питание нервной ткани: актовегин, трентал, церебролизин и другие.
Паллиативная терапия направлена на облегчение состояние тяжелобольных пациентов.
Ученые всего мира пытаются создать препарат для лечения патологии. Известно, что уже разработано несколько препаратов, которые возможно сумеют помочь пациентам. Они находятся на стадии клинических испытаний и их эффективность и безопасность для организма только изучается.
Родителям детей с тяжелым, неподдающимся лечению, диагнозом, для рождения здорового ребенка необходимо планировать беременность с репродуктологом. Искусственное оплодотворение, использование донорской спермы или яйцеклетки, экстракорпоральное оплодотворение – одни из способов родить здорового ребенка носителям дефектного гена.
Прогноз: сколько живут пациенты?
Спинальная амиотрофия крайне тяжелое заболевание, которое существенно ухудшает качество жизни больных и является одной из причин младенческой смерти. Пациенты с таким диагнозом являются инвалидами и зачастую не в силах самостоятельно себя обслуживать.
Многие пациенты не могут ходить и стоять, передвигаясь с помощью инвалидной коляски. При поражении рук больные не способны делать элементарных вещей: есть, держать небольшие предметы, читать, умываться и прочее. Они нуждаются в дополнительном уходе и постоянном медицинском наблюдении. При тяжелом течении они дышать лишь с помощью аппарата ИВЛ и питаются через зонд.
Продолжительность жизни во многом зависит от формы заболевания. Наиболее тяжелой является амиотрофия Верднига, при которой больные дети редко достигают 4 лет. Пациенты со вторым типом болезни редко доживают до совершеннолетия. Наиболее благополучным являются второй и третий тип болезни, при которых пациенты достигают зрелого возраста.
В настоящее время спинальная амиотрофия относится к неизлечимым заболеваниям, часто приводит к летальному исходу.
Спинальная амиотрофия (спинальная мышечная атрофия или СМА) — это неизлечимое, почти всегда наследуемое заболевание, вызванное мутацией гена в 5-й хромосоме.
Мутация гена СМА приводит к недостатку белка, который необходим для построения белковых РНК-структур, и к недостаточному развитию поперечнополосатых мышц, преимущественно нижних конечностей, а также шейного отдела и головы.
Болезнь может проявиться с момента рождения, и даже когда плод еще в утробе, и в любой период жизни. У новорожденных спинальная амиотрофия чаще всего приводит к ранней смерти, но в некоторых случаях она может иметь мягкие формы, в основном в пожилом возрасте. Рассмотрим подробнее особенности этой патологии.

Спинальная амиотрофия — все о болезни
СМА — достаточно редкое заболевание, открытое немецким врачом Верднигом в 1891 г. Заболевает ею один человек из 6 — 10 тыс., однако носителем рецессивного гена СМА является каждый 50-й человек.
В 1898 г. Вердниг и еще один ученый Гофман установили, что причиной СМА является дегенеративное поражение и недостаточное количество двигательных (моторных) нейронов передних рогов спинного мозга — SMN (survival motor neurons).
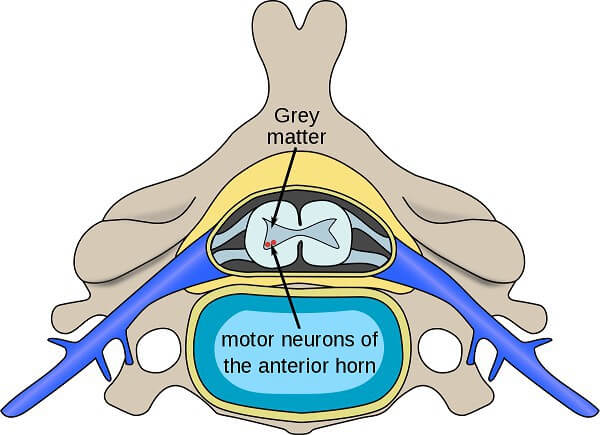
Уже в 20-м веке (в 1956 г.) другие ученые Кугельберг и Веландер открыли менее злокачественную, с более мягкими проявлениями, форму СМА, которой болеют в ювенильном и взрослом возрасте.
Болезнь может наследоваться по любому из типов:
- аутосомно-доминантному;
- аутосомно-рецессивному;
- Х-сцепленному доминантному;
- Х-сцепленному рецессивному.
В связи с этим, классифицируется очень много разных форм СМА.
Детская форма СМА наследуется по аутосомно-рецессивному типу: если оба родителя — носители, то заболеет четвертая часть их потомства.
Аутосомно-доминантный тип наследования СМА приводит к проявлению болезни у детей с вероятностью 50%, даже если болен всего один родитель.
Подробнее о сути наследования можно прочитать в статье о синдроме Марфана.
Спинальная мышечная атрофия разделяется на четыре формы:
- Младенческая (I) — спинальная мышечная атрофия Верднига-Гофмана: диагностируется с момента рождения до полугода.
- Промежуточная (II) — болезнь Дубовица: от семи месяцев до полутора лет.
- Юношеская (III) — б. Кюгельберга-Веландера: после полутора лет.
- Взрослая (IV): после 35 лет.
Общие симптомы при СМА:
- поражение проксимальных (средних) мышц и фасций;
- сохранение чувствительности в большинстве клинических случаев;
- задержки умственного и психического развития при спинальной мышечной амиотрофии крайне редки;
- возможна при некоторых видах атрофия не только мышц конечностей, но респираторных, жевательных и глотательных.
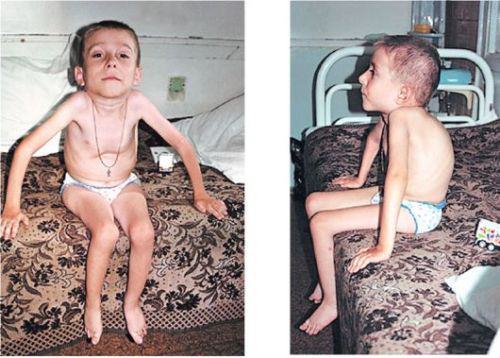
- Самой тяжелой и неблагоприятной считается СМА первого вида (младенческая) — спинальная амиотрофия Верднига — Гоффмана, при которой дети не способны совершать активные движения, держать голову, самостоятельно сидеть. С большим трудом дается младенцу кормление, так как ему трудно сосать молоко и глотать его.
- Меньшей злокачественностью обладает болезнь Дубовица (II промежуточная форма СМА): при ней дети способны сидеть, держать голову и есть, однако ходить всё же неспособны.
- Наименее тяжелой является юношеская форма: несмотря на мышечную слабость, ребенок способен научиться ходить, однако заболевание хоть и медленно, но прогрессирует и может привести к ранней инвалидности.
- Четвертая взрослая форма СМА может привести из-за слабости м — ц проксимальных отделов к невозможности самостоятельного передвижения, выпадению рефлексов, но прогноз в отношение длительности жизни при этом остается благоприятным.

Кроме мутации в генах, вызывающих поражение проксимальных мышц, существуют подобные патологии, с разным типом наследования, приводящие к атрофии мышц и фасций дистальных (концевых отделов).
Список их довольно велик, сведем болезни в небольшую таблицу:
| Название СМА | Тип наследования | Особенности и симптомы |
| SMAX1 | Х -сцепленный рецессивный | Наблюдается в основном у пожилых, поражает бульбарные нервы черепа, вызывает нисходящий паралич. |
| SMAХ2 | Х — сцепл. рецессивный | Врожденная агрессивная форма, приводящая к смерти до 3 — х мес. Вызывает слабость, арефлексию, контрактуры и переломы. |
| SMAX3 | Х — сцепл. рецессивный | Поражает в основном мальчиков. Атрофия всех дистальных мышц. Медленное нарастание симптомов |
| Дистальная ДСМА1 | аутосомно — рецессивный | Врожденная, поражаются в основном руки, возможны тяжелые дыхательные нарушения |
| Дистальные формы ДСМА2 — ДСМА5 | аутосомно — рецессивный | Все четыре формы отличаются медленным прогрессированием, ДСМА5 диагностируется у молодых. |
| Дистальная СМА двух типов: VA и VB (DSMAVA и DSMAVB) | аутосомно-доминантный | Преимущественно атрофированы верхние конечности. |
| ДСМА тип 2D | аутосомно — рецессивный | Юношеское и взрослое заболевание с медленным развитием: поражаются и проксимальные, и дистальные м — цы вначале в голенях, затем в руках. |
| ДСМА тип 7А | аутосомно-доминантный | Очень редкая взрослая форма с поражением голосовых связок. |
| ДСМА тип 2А | аутосомно-доминантный | Разновидность болезни Шарко (аллельный тип) |
| Ювенильная SMA (тип HMN1) | аутосомно-доминантный | Встречается в юности |
| Врожденная спинальная амиотрофия | аутосомно-доминантный | Нарушение иннервации и атрофия м — ц бедер, стоп, коленей с контрактурой и деформацией; иногда поражается голосовые связки. |
| SMA Финкеля | аутосомно-доминантный | Начинается преимущественно в 35 — 37 лет, но зафиксированы случаи заболевания и в детском возрасте. Медленно развивается вначале в ногах, а затем в руках. Активность и рефлексы снижены, наблюдается непроизвольное дрожание (фасцикуляция). |
| SMA Джокела | аутосомно-доминантный | Поражаются у взрослых прокс. и дистальные м — цы. |
| SMA (тип LED1) | аутосомно-доминантный | Атрофия нижних конечностей у новорожденных. |
| SMA типа РМЕ | аутосомно — рецессивный | Атрофия дистальных мышц с нарушением иннервации и эпилептическими припадками |
| СМА с врожденными костными переломами | аутосомно — рецессивный | Тяжелые симптомы, как при болезни. Верднига-Гоффмана, отягощенные переломами. |
| СМА с гипоплазией | аутосомно-доминантный | Врожденная аномалия головного мозга с церебральными симптомами, микроцефалией и задержкой развития. |
| СМА ювенильная асимметричного типа | -------------- | Ею болеют молодые индийские мужчины |

В этой таблице следует обратить внимание на последние два вида спинальной амиотрофии:
- СМА с гипоплазией сопровождается отклонениями умственного и психического развития, что не характерно для остальных видом болезни.
- Асимметричная ювенильная (индийская) амиотрофия не передается по наследству. При этом заболевание после двух-пяти лет вялого течения может стабилизироваться. Симптомы фасцикуляции при этой специфической форме наблюдаются редко.
Лечение спинальной амиотрофии
Вылечить подобного рода болезни, как и любую наследственную патологию, затрагивающую спинной или головной мозг, кардинально невозможно. Эффективность многих используемых сегодня лекарств при терапии амиотрофии не доказана. Суть лечения сводится к увеличению белка, участвующего в формировании двигательных нейронов SMN.
Так, в основном применяются следующие препараты:
- вальпроевая кислота;
- оксибутират натрия;
- нусинерсен (новое лекарство, введенное в США в 2016 г. для лечения СМА).
Также необходимы поддерживающее лечение (ЛФК, массаж, физиотерапия), специальная белковая диета, при которой учитываются возможные противопоказания. Больные, лишенные возможности самостоятельного передвижения, нуждаются в социальной опеке.

Профилактика спинальной амиотрофии возможна лишь в плане раннего выявления мутации в эмбриональном периоде: проводится ДНК-анализ при помощи специальной биопсии. Положительный результат анализа может быть основанием для прерывания беременности, если так решит мать.
Читайте также:
