
Что такое спинальная амиотрофия взрослых
Спинальная мышечная атрофия является основной генетической причиной смерти в детском возрасте. Давайте разберемся в причинах и способах изменения жизни ребенка, а также узнаем, какие методы лечения доступны на сегодняшний день.
Что такое спинальная мышечная атрофия
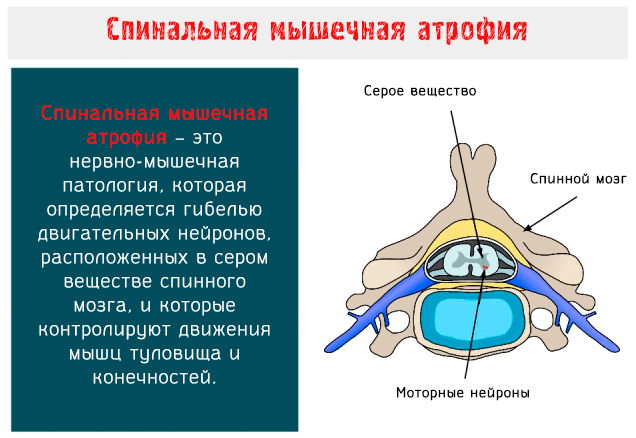
Спинальная мышечная атрофия (SMA: Spinal Muscular Atrophy) – это нервно-мышечное заболевание аутосомно-рецессивного типа, характеризуется гибелью двигательных нейронов, расположенных в переднем роге серого вещества спинного мозга и в нижней части ствола головного мозга.
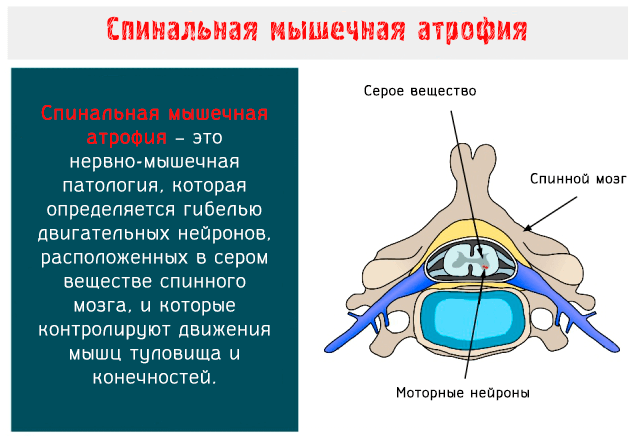
Моторные нейроны – это клетки, из которых образуются нервы, предназначенные для управления скелетными и поперечно-полосатыми мышцами глотки и гортани: когда они вырождаются, целые группы волокон подвергаются атрофии и, соответственно, результатом является мышечная слабость.
Работы глазных мышц, хотя и управляется моторными нейронами энцефального ствола мозга, не нарушается при этой болезни.
Частота спинальной мышечной атрофии колеблется от 1:6000 до 1:10000, и подвержены ей все этнические группы; является редким заболеванием, является одним из самых распространенных нервно-мышечных заболеваний, точнее вторым после дистрофии Дюшенна.
Причина спинально мышечной атрофии
Причина спинально мышечной атрофии была обнаружена в середине 90-х годов, спустя сто лет после первого описания болезни. В 95% случаев речь идёт о делеции в гене SMN1, локализованном на длинном плече хромосомы 5 (делеция – это потеря последовательности ДНК).
Поскольку спинально мышечная атрофия наследуется по аутосомно-рецессивному типу, для развития болезни человек должен получить обе копии плохого SMN1 – от матери и от отца. Таких родителей называют гетерозиготными или носителями, и они не имеют симптомов заболевания. Носители встречаются с частотой 1:50.
Ген SMN1 кодирует белок SMN, который используется в цитоплазме и ядре всех клеток и имеет решающее значение для формирования snRNP, малых ядерных рибонуклеопротеидов, компонентов сплайсинг машин.
Почему же вездесущий белок SMN является критическим фактором для выживания и надлежащего функционирования моторных нейронов?
В 2012 году Лотти и соавторы показали, что белки SMN имеют имеют важное значение для дифференциации и бесперебойной работы моторных нейронов.
Другие гипотезы были сформулированы для объяснения антиапоптической роли SMN:
- потребность в этом белке выше у моторных нейронов, чем в других тканях.
- по мнению других авторов, это можно объяснить тем, что белок SMN участвует в транспортировке вдоль аксонов РНК-связывающих белков.
Несмотря на все предположения, в настоящее время ещё неясно, какая из многих функций протеина SMN связана с развитием спинально мышечной атрофии.
4 типа спинально мышечной атрофии
Спинально мышечную атрофию классифицируют на четыре типа, в соответствии:
- с возрастом появления симптомов
- с максимальной двигательной активностью, на которую способен больной
У 25% лиц избегают точной классификации. Кроме того, у людей, страдающих от одного типа заболевания, симптомы могут существенно различаться.
Это самая тяжелая форма спинально мышечной атрофии, составляет 50% от всех случаев.
Главные её особенности:
- проявляется до 6-го месяца жизни
- ребенок имеет плохую и дряблую мышечную массу: он мало движется, потому что не может противостоять силе тяжести, не в состоянии держать голову в вертикальном положении и сидеть без поддержки
- кости хрупкие и подвержены переломам, кроме того, в позвоночнике развивается сколиоз. Проблемы с костями у пациента со спинально мышечной атрофией не удивляют, так как именно физическая активность способствует минерализации костей
- рефлекс сосания и глотания слабый, поэтому такого ребёнка трудно кормить
- грудная клетка ребенка меньше нормы из-за слабости дыхательных мышц. Кашлевый рефлекс слабый, что нарушает процесс избавления от выделений (слизи и твердые частицы, включая микробов)
У детей, страдающих от спинально мышечной атрофии 1 типа, часто развивается пневмония, так как они не в состоянии избавиться от каких-либо патогенных микроорганизмов с кашлем, а также из-за потери контроля глотательных мышц, которые не могут предотвратить попадание слюны и кусочков пищи в легкие. Повторяющие пневмонии ведут, к сожалению, к дыхательной недостаточности.
Для тех, кто страдает от этой формой патологии прогноз неблагоприятный: смерть наступает в течение 2 лет, даже самое хорошее лечение продлевает жизнь только до 5 лет.
Промежуточная форма спинальной мышечной атрофии.
Давайте посмотрим характеристики:
- проявляется между 6 и 18 месяцами
- ребенок показывает задержку в развитии моторики: не в состоянии сидеть, ему нужна поддержка, чтобы стоять, и никогда не научится ходить. Может иметь легкий тремор рук
- при этом типе также отмечается склонность к развитию сколиоза и хрупкости костей
- у некоторых маленьких пациентов дисфагия становится препятствием для поглощения достаточного для развития количества калорий
- кашлевый рефлекс может ослабнуть, облегчая возникновение респираторных инфекций
При спинальной мышечной атрофии 2 типа также высок риск развития дыхательной недостаточности. Прогрессирование симптомов настолько разнообразно, что некоторые пациенты умирают в младенчестве, другие в состоянии достичь зрелости.
Детская форма спинальной мышечной атрофии, которая:
- может возникнуть в возрасте от полутора лет
- по сравнению с предыдущими случаями, дети могут стоять и ходить самостоятельно, эта способность в некоторых случаях сохраняется до зрелого возраста
- наблюдается тремор рук и могут возникнуть проблемы с суставами и сколиоз
- нарушения дыхания и глотания проявляются менее часто, чем при 1 и 2 типе
У людей, страдающих от 3 типа спинальной мышечной атрофии, средняя продолжительность жизни сравнима со здоровыми людьми. Но, из-за проблем с питанием и низкой физической активность, часто имеют избыточный вес.
Продолжительность жизни нормальная.
Как распознать спинальную мышечную атрофию
Специалист по детской неврологии задаст ряд вопросов, чтобы получить подробный отчет о медицинской истории ребенка и его семьи, после процедуры физического обследования, чтобы оценить физическое состояние маленького пациента.
Подтверждение диагноза спинальной мышечной атрофии достигается благодаря генетическому тесту: берут образец крови и исследуют на наличие аномального гена SMN1. Тест можно использовать, чтобы найти носителей.
Поиск неисправного SMN1 также может осуществляться путём биопсии ворсинок хориона, которые являются частью плаценты, что делает возможным пренатальную диагностику в случае:
- если у пары уже был ребёнок, пострадавший от спинальной мышечной атрофии
- партнеры обнаруживают, что являются носителями, но все равно хотя родить ребёнка
Иногда бывает, что нельзя точно утверждать, что это спинальная мышечная атрофия. Тогда используют другие тесты, которые помогают провести дифференциальный диагноз между спинальной атрофией и другими патологиями нервов и мышц:
- электромиография, которая измеряет электрическую активность мышц
- мышечная биопсия, то есть изучение образцов мышечной ткани
- оценка концентрации креатина киназы, фермента уровень которого повышается при повреждении мышц
Как облегчить симптомы спинальной атрофии
На данный момент не существует лекарств для лечения спинальной мышечной атрофии, поэтому пациенты могут воспользоваться только поддерживающим лечением.
- физиотерапия
- диетология
- дыхание
Для пациентов школьного возраста важно, чтобы они активно участвовали в школьных мероприятиях, потому что их физическая инвалидность никак не влияет на способности к обучению.
Физиотерапия необходима независимо от возраста человека. Упражнения позволят максимизировать амплитуду движений, чтобы предотвратить или замедлить потерю мелкой моторики. Дети со спинальной мышечной атрофией 1 и 2 типа получают огромную пользу от гимнастики в бассейне, поскольку вода помогает стимулировать всю мышечную массу.
Пациентам с 3 типом спинальной мышечной атрофии нужны ортопедические устройства (инвалидные коляски, параподы и т.д.), которые обеспечивают удобство и мобильность. Упражнения также важны, поскольку помогают предотвратить сколиоз, который усугубляет проблемы с дыханием и движениями.
Каждый человек, страдающий от спинальной мышечной атрофии, должен иметь свой индивидуальный план питания, чтобы предотвратить последствия недостаточного или избыточного питания.
У тех детей, которые имеют большие трудности при грудном кормлении, пережевывании пищи и глотании, нужно принять меры, чтобы избежать таких осложнений, как аспирационная пневмония.
- Вы можете прибегнуть к использованию назогастрального зонда, который проходит через нос и доставляет пищу в желудок. Его относительно легко установить и снять, но он может протекать, тогда его следует заменить
- Другой вариант – гастростомия, то есть вывод трубки из желудка; является более простым в обслуживании, но процедура выполняется в операционной под наркозом.
Дыхание
Есть в этом случае для со спинальной мышечной атрофией существует три цели:
- пациенты и всех люди, которые вступают в контакт с ними, должны быть вакцинированы, например, против вируса гриппа, пневмококковой инфекции и бактерии коклюша, потому что инфекции дыхательных путей могут быть очень опасными для таких пациентов
- если кашлевый рефлекс слабым, это можно исправить с помощью специального устройства (Cough Assist): оно создает быстрое изменение давления снаружи и внутри легких, и быстрое прохождение воздуха по дыхательным путям, что имитирует кашель, освобождая дыхательные пути от секрета и микробов
- наконец, важна оценка дыхательной функции этих субъектов по степени сатурации кислорода в крови. Если количества кислорода меньше потребностей, стоит серьёзно рассмотреть идею использования механического респиратора. Изначально он используется в случае инфекции дыхательных путей и во время сна; с развитием атрфоии – весь день.
Открытие причины заболевания открыло для исследовательских групп большое направление для поиска методов лечения, направленных на максимально возможное замедление прогрессирования симптомов: повышение уровня белка SMN.
- Поскольку спинальная мышечная атрофия является моногенным заболеванием, это позволяет вмешаться в корень недуга, предоставляя пациентам функционирующий ген SMN1 (генная терапия)
- У лиц, страдающих от спинальной атрофии, но имеющих ген SMN2, можно увеличить экспрессию этого гена и заблокировать исключение экзона 7 во время сплайсинга незрелой мРНК.
В обоих случаях количество функционирующего белка SMN увеличивается.
AVXS-101 – экспериментальный препарат, разработанный биотехнологической компании Авексис, которому удалось достичь 1 этапа экспериментов на людях, при оценке безопасности лечения, и он начинает проверку эффективности.
Авексис сосредоточились на детях, страдающих от спинальной мышечной атрофии 1 типа, потому что это самый распространенный и смертельный тип заболевания.
AVXS-101 состоит из большого числа частиц адено-ассоциированного вируса серотипа 9, неспособного к репликации, но содержащего одну копию нормального гена SMN1.
Вводится в организм внутривенно, в состоянии преодолеть гематоэнцефалический барьер и достичь моторных нейронов.
Молекула ДНК, переносимая каждым вирусным вектором, производится в лаборатории. Она не изменяет ДНК пациента; содержит промотор, т.е. последовательность, которая способствует транскрипции ДНК в РНК, и гарантирует постоянное производство протеина SMN.
Анализ промежуточных данных, опубликованных Авексис в апреле 2016 года показывает, что:

Спинальные амиотрофии — это генетические заболевания, проявляющиеся мышечной атрофией и обусловленные дегенеративными изменениями спинальных мотонейронов и моторных ядер ствола головного мозга. Общим симптомокомплексом выступают симметричные вялые параличи с атрофиями мышц и фасцикуляциями на фоне интактной чувствительной сферы. Диагностируются спинальные амиотрофии по данным семейного анамнеза, неврологического статуса, ЭФИ нервно-мышечного аппарата, МРТ позвоночника, ДНК-анализа и морфологического исследования мышечного биоптата. Лечение малоэффективно. Прогноз зависит от формы спинальной мышечной атрофии и возраста ее дебюта.

- Причины
- Патогенез
- Классификация
- Симптомы спинальных амиотрофий
- Болезнь Верднига-Гоффмана
- Ювенильная спинальная амиотрофия Кугельберга-Веландера
- Бульбоспинальная амиотрофия Кеннеди
- Дистальная СМА Дюшенна-Арана
- Скапуло-перонеальная СМА Вюльпиана
- Осложнения
- Диагностика
- Дифференциальная диагностика
- Лечение спинальных амиотрофий
- Немедикаментозная терапия
- Медикаментозная терапия
- Хирургическое лечение
- Экспериментальное лечение
- Прогноз
- Профилактика
- Цены на лечение
Общие сведения
Спинальные амиотрофии (спинальные мышечные атрофии, СМА) — наследственно обусловленные заболевания, в основе которых лежит дегенерация мотонейронов спинного мозга и ствола головного мозга. Описаны в конце XIX века. Их частота составляет 1 случай на 6-10 тыс. новорожденных. Около 85% спинальных мышечных атрофий составляют проксимальные формы с более выраженной слабостью и атрофиями проксимальных мышечных групп конечностей. На долю дистальных форм приходится лишь 10% СМА. На сегодняшний день спинальные амиотрофии представляют практический интерес для целого ряда дисциплин: детской и взрослой неврологии, педиатрии, генетики.
Причины
Благодаря современной генетике установлено, что возникающие дегенеративные процессы двигательных нейронов обусловлены мутациями в генах SMN, NAIP, H4F5, ВTF2p44, расположенных на 5-ой хромосоме в локусе 5q13. Несмотря на то, что спинальные амиотрофии детерминируются аберрациями одного хромосомного локуса, они представляют собой группу разнородных нозологий, одни из которых проявляются в младенческом возрасте, а другие манифестируют у взрослых. В большинстве случаев амиотрофии наследуются аутосомно-рецессивно.
Патогенез
Генетические мутации приводят к развитию дегенеративных изменений в передних рогах спинного мозга. Нарушается иннервация и нейротрофика поперечно-полосатой мускулатуры. В результате постепенно возникает атрофия мышечной ткани. Преимущественное поражение отдельных групп мышц (проксимальных или дистальных частей верхних или нижних конечностей) у различных форм спинальных амиотрофий отличается. Характерно отсутствие нарушений чувствительности.
Классификация
Общепринятым считается разделение спинальных мышечных атрофий на детские и взрослые. Детские СМА классифицируются на ранние (дебютирующие в первые месяцы жизни), более поздние и ювенильные. Детские спинальные амиотрофии представлены:
- амиотрофией Верднига-Гоффманна;
- ювенильной формой Кугельберга-Веландера;
- хронической инфантильной СМА;
- синдромом Виалетто-ван Лэре (бульбоспинальная форма с глухотой);
- синдромом Фацио-Лонде.
Взрослые формы СМА манифестируют в возрасте от 16 до 60 лет и отличаются более доброкачественным клиническим течением. К СМА взрослого возраста относятся:
- бульбоспинальная амиотрофия Кеннеди;
- скапулоперонеальная;
- лицелопаточноплечевая и окулофарингеальная формы;
- дистальная СМА;
- мономелическая СМА.
Выделяют также изолированные и сочетанные спинальные амиотрофии. Изолированные СМА характеризуются преобладанием поражения спинальных мотонейронов, которое во многих случаях является единственным проявлением заболевания. Сочетанные спинальные амиотрофии представляют собой редкие клинические формы, при которых симптомокомплекс амиотрофии комбинируется с другой неврологической или соматической патологией. Описаны сочетания СМА с врожденными пороками сердца, глухотой, олигофренией, понтоцеребеллярной гипоплазией, врожденными переломами.
Симптомы спинальных амиотрофий
Общим для спинальных мышечных атрофий является симптомокомплекс симметричного вялого периферического паралича: слабость, атрофия и гипотония мышечных групп одноименных конечностей (чаще вначале обеих ног, а затем и рук) и туловища. Пирамидные нарушения не типичны, но могут развиваться на поздних стадиях. Расстройства чувствительности отсутствуют, функция тазовых органов сохранена. Обращает внимание более выраженное поражение проксимальных (при проксимальных СМА) или дистальных (при дистальных СМА) мышечных групп. Типично наличие фасцикулярных подергиваний и фибрилляций.
Встречается в 3-х клинических вариантах. Врожденный вариант дебютирует в первые 6 мес. жизни и является наиболее злокачественным. Его симптомы могут проявляться еще во внутриутробном периоде слабым шевелением плода. Дети с рождения имеют мышечную гипотонию, не способны переворачиваться и держать голову, при более позднем дебюте — не могут сидеть. Патогномонична поза лягушки — ребенок лежит с разведенными в стороны и согнутыми в коленях и локтях конечностями.
Амиотрофии имеют восходящий характер — вначале возникают в ногах, затем вовлекаются руки, позже — дыхательная мускулатура, мышцы глотки и гортани. Сопровождается задержкой психического развития. К 1,5 годам наступает смертельный исход.
Ранняя спинальная амиотрофия манифестирует до 1,5 лет зачастую после инфекционного заболевания. Ребенок утрачивает двигательные способности, не может стоять и даже сидеть. Периферические парезы сочетаются с контрактурами. После вовлечения дыхательных мышц развивается дыхательная недостаточность и застойная пневмония. Летальный исход обычно происходит в возрасте до 5-ти лет. Поздний вариант дебютирует после 1,5 лет, отличается сохранением двигательной способности до 10-летнего возраста. Летальный исход наступает к 15-18 годам.
Характеризуется дебютом в период от 2 до 15 лет. Начинается с поражения проксимальных мышц ног и тазового пояса, затем захватывает плечевой пояс. Около четверти пациентов имеют псевдогипертрофии, что делает клинику сходной с проявлениями мышечной дистрофии Беккера. В плане дифдиагностики большое значение имеет наличие мышечных фасцикуляций и данные ЭМГ. Течение амиотрофии Кугельберга-Веландера доброкачественное без костных деформаций, в течение ряда лет пациенты остаются способными к самообслуживанию.
Наследуется рецессивно сцеплено с Х-хромосомой, манифестирует только у мужчин после 30-летнего возраста. Типично медленное, относительно доброкачественное течение. Дебютирует с амиотрофии проксимальных мышц ног. Бульбарные расстройства появляются через 10-20 лет и благодаря медленному прогрессированию не вызывают нарушения витальных функций. Может наблюдаться тремор головы и рук. Патогномоничным симптомом выступают фасцикулярные подергивания в периоральных мышцах. Зачастую отмечается эндокринная патология: атрофия яичек, снижение либидо, гинекомастия, сахарный диабет.
Осложнения
К наиболее частым неблагоприятным последствиям можно отнести постоянные падения и ассоциированные с ними патологические переломы, что связано с постепенно нарастающей мышечной атрофией мышц нижних конечностей. При бульбоспинальной амиотрофии Кеннеди нередко наблюдаются осложнения со стороны эндокринной и репродуктивной систем – эректильная дисфункция, первичное бесплодие, сахарный диабет.
Более тяжелые состояния встречаются реже, в основном при болезни Верднига-Гоффмана или на поздних стадиях других амиотрофий. Вследствие выраженной атрофии мышц глотки и дыхательной мускулатуры пища попадает в дыхательные пути (аспирация), развивается дыхательная недостаточность. Возможны грубые костно-суставные деформации, утрата способности к ходьбе и самообслуживанию. В единичных случаях обнаруживается рак грудной железы.
Диагностика
Больных со спинальными амиотрофиями курируют врачи-неврологи, а при манифестации в детском возрасте – педиатры или неонатологи. При необходимости может потребоваться привлечение для консультации других специалистов – эндокринологов и генетиков. Для диагностики некоторых разновидностей амиотрофий большое значение имеют анамнестические данные, а именно возраст появления симптоматики (например, болезнь Верднига-Гоффмана всегда дебютирует у детей до 6 месяцев, а амиотрофия Кугельберга-Веландера – после 2 лет).
Во время осмотра пациента обращается внимание на снижение общего мышечного тонуса, ослабление или утрату сухожильных рефлексов, скелетно-мышечные деформации. У части больных отмечается псевдогипертрофия икроножных мышц. Чтобы подтвердить или исключить диагноз назначается следующее дополнительное обследование:
Дифференциальный диагноз спинальных амиотрофий необходимо проводить с другими наследственными нейродегенеративными заболеваниями, поражающими мышечную ткань. К таким патологиям можно отнести:
Лечение спинальных амиотрофий
Все без исключения больные должны быть госпитализированы в стационар. В тяжелых ситуациях (например, при дыхательной недостаточности вследствие слабости дыхательных мышц) пациентов переводят в отделение реанимации и интенсивной терапии и подключают к аппарату ИВЛ. Все мероприятия направлены на облегчение состояния пациента. Несмотря на это, комплексный подход и строгое соблюдение рекомендаций врачей позволяют улучшить качество жизни больного. Виды консервативной терапии, применяющиеся для терапии спинальных амиотрофий:
- Обеспечение питания. При значительном нарушении акта глотания особое внимание уделяется вопросу кормления. Консистенция пищи должна быть полутвердой, положение пациента вертикальным. Может понадобиться установка назогастрального зонда.
- ЛФК. С целью повышения тонуса мышц и замедления их атрофии рекомендуются регулярные физические нагрузки. Наиболее эффективно совмещение активных (выполняются специалистом) и пассивных упражнений (выполняются самим пациентом).
- Физиолечение. Для активации метаболизма в мышечных тканях назначаются сеансы электростимуляции модулированным током, грязевые аппликации, электрофорез.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах выполняются различные виды массажа – ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Ортопедическое лечение. Для предупреждения и коррекции костных деформаций и суставных контрактур рекомендуется использование ортопедических приспособлений: корсетов, ортезов, ортопедической обуви.
- Респираторная поддержка. Нередко возникает необходимость в устранении кислородной недостаточности. В зависимости от тяжести состояния больного назначаются ингаляции кислорода через лицевую маску/назальную канюлю или неинвазивная вентиляция легких через портативные аппараты ИВЛ.
Для достижения максимального эффекта лечение должно проводиться непрерывно и подбираться индивидуально для конкретного пациента.
Этиотропная терапия с доказанной эффективностью на сегодняшний день отсутствует. Лекарственные препараты, применяющие для лечения спинальных амиотрофий, следующие:
- Метаболические средства. Для улучшения обменных процессов в мотонейронах и мышечных тканях применяются препараты, стимулирующие функцию митохондрий (коэнзим Q10, янтарная кислота), ноотропы (пирацетам, гамма-аминомасляная кислота), L-карнитин.
- Вальпроаты и Кленбутерол. Исследования показали, что противоэпилептические препараты из группы производных вальпроевой кислоты и агонист бета-адренергических рецепторов Кленбутерол способны увеличивать образование белка выживаемости мотонейронов (SMN) и, соответственно, улучшать клиническое течение заболевания.
- ИПП и прокинетики. Пациентам с гастроэзофагальным рефлюксом, который нередко развивается при нарушении глотания, назначаются ингибиторы протонной помпы (эзомепразол) и средства, стимулирующие моторику желудочно-кишечного тракта (итоприд).
- Муколитики и отхаркивающие средства. У больных со слабостью дыхательной мускулатуры для устранения таких проблем как слабое отхаркивание и скопление в дыхательных путях густой мокроты применяются препараты, разжижающие мокроту (ацетилцисетин) и стимулирующие ее отхаркивание (терпингидрат).
- Гормональные препараты. Людям с возникшими эндокринологическими осложнениями показаны инсулин, сахароснижающие препараты (метформин), тестостерон и антиандрогены (дутастерид).
При развитии грубых деформаций грудной клетки и позвоночника или крайне выраженных контрактур суставов показаны ортопедические операции. Лежачим больным, страдающим постоянно рецидивирующими пневмониями, выполняется трахеостомия. При гастроэзофагеальном рефлюксе, резистентном к медикаментозному лечению, прибегают к лапароскопической фундопликации Ниссена.
Постоянно ведутся многочисленные исследовательские работы по разработке лекарственных средств, способных остановить или хотя бы замедлить процесс нейродегенерации. Значительные успехи достигнуты в области генной терапии. Уже используются в клинической практике препараты, корректирующие дефекты матричной РНК гена SMN – Спинраза и Рисдиплам. В конце 2019 года был зарегистрирован и одобрен к клиническому применению препарат Золгенсма, который содержит функционально полноценный ген SMN1.
Прогноз
Прогноз всецело зависит от клинического варианта СМА и возраста ее манифестации. Наиболее неблагоприятный прогноз имеют детские спинальные амиотрофии, при начале в младенческом возрасте они зачастую приводят к летальному исходу в течение первых 2-х лет жизни ребенка. Спинальные амиотрофии взрослого возраста отличаются способностью больных самостоятельно обслуживать себя в течение многих лет, а при медленном прогрессировании имеют благоприятный прогноз не только для жизни, но и для трудоспособности пациентов (при создании для них оптимальных условий труда).
Профилактика
Специфических методов первичной профилактики не существует. Единственный способ предотвратить возникновение болезни – пренатальная диагностика (обнаружение мутаций в ворсинах хориона или амниотической жидкости) с прерыванием беременности. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
Спинальная мышечная атрофия – одно из самых опасных генетически обусловленных заболеваний, которое обнаруживается у младенцев, подростков, взрослых.

Страшно узнать, что малыш никогда не будет сидеть, стоять, бегать. Еще страшнее видеть, как нормально растущий и развивающийся ребенок вдруг начинает медленно угасать, постоянно падать, через несколько месяцев не может подняться по лестнице, а однажды теряет способность просто встать.
Спинальная мышечная атрофия — что это
Врачи объединяют несколько видов наследственных заболеваний, характеризующихся нарушением движения, в одну группу под названием спинальная мышечная атрофия. В МКБ-10 они идут под кодом G12 с дополнительными указаниями на тип болезни.
Спинальная мышечная атрофия — это разнородная группа наследственных заболеваний, протекающих с поражением / потерей двигательных нейронов передних рогов спинного мозга.
По данным исследователей, около 0,01-0,02% детей рождаются с диагнозом СМА. Чаще патология встречается у мальчиков и мужчин.
Обнаруживается спинальная мышечная атрофия преимущественно у детей в раннем возрасте. Однако некоторые формы заболевания начинают проявляться только у подростков или уже взрослых людей. Коварство патологии заключается в том, что она постепенно, день за днем отбирает у больных то, что они сумели добиться.
Впервые патологию описал Г. Вердниг. Он обратил внимание на равностороннюю атрофию спинного мозга, его передних рогов, корешков периферических нервов в 1891 г. Уже в следующем году Дж. Хоффман сумел доказать, что речь идет о самостоятельном заболевании. В середине XX в. исследователи Е. Кугелберг и Л. Веландер описали патологию, которая возникает в позднем возрасте и имеет более благоприятный прогноз.
Симптомы
Каждый вид СМА имеет свои особенные признаки, однако существуют некоторые симптомы, которые позволяют объединить разнородные заболевания в одну группу. Это:
- Нарастающая слабость мышц и их атрофия.
- При заболевании, проявившемся после 1-2 лет, заметна деградация уже достигнутых способностей, например, бега, ходьбы.
- Тремор пальцев. Дрожь наблюдается и на языке.
- Деформация скелета.
- Сохранность интеллектуального и психического здоровья у большинства больных.

Виды СМА
Возраст, время проявления симптомов, особенности течения патологии, прогноз позволяют выделять несколько видов заболеваний.
Данная форма патологии описывается редко, часто его объединяют с первым типом СМА. Болезнь – врожденная. Характеризуется полным отсутствием движений, сухожильных рефлексов, слабостью мышц, ограниченным движением суставов коленей. С самого рождения наблюдаются дыхательные нарушения.
Часто диагноз путают с перинатальной энцефалопатией или родовыми травмами. Однако в последних двух случаях дети достаточно быстро адаптируются, их состояние становится лучше. У детей со СМА улучшения не возникают, в большинстве случаев они умирают, не дожив до месяца, от осложнений.
Патология первого типа имеет очень тяжелое течение. Ее называют также болезнью Верднига-Гоффмана. Диагностирован этот тип может быть от рождения до 6 месяцев. Отмечается слабость мышц, их периодическое подергивание – последнее увидеть достаточно трудно из-за достаточно большого слоя жирового слоя. Дрожь может периодически пробегать по языку малыша.
Наблюдается ухудшение рвотного, сосательного, глотательного рефлекса, нарушение слюноотделения. Младенец не может кашлять, громко кричать. Часто сопровождается тяжелыми дыхательными нарушениями, пневмонией.
Грудная клетка у таких детей имеет более плоскую форму из-за слабо развитых мышц груди.
Малышей со спинальной амиотрофией Верднига-Гоффмана легко узнать по позе лягушонка. Бедра и плечи отведены, локти и колени согнуты.
К 6 месяцам ребенок может научиться держать головку, но практически никогда не сможет самостоятельно сесть, встать, ходить. Проблемы с глотанием вызывают сложности в кормлении.
Часто именно это заболевание сопровождается олигофренией, врожденными нарушениями работы сердца, небольшим размером головы.
Патология второго типа обнаруживается у малышей в возрасте от полугода до полутора-двух лет. Болезнь Дубовица характеризуется слабостью и тремором в глубоких отделах мышц, дрожью пальцев, языка, ограничением объема движения конечностей. Детей отличает маленький вес, задержка развития. Они сидят, сами кушают, но вставать и ходить не могут.
Болезнь носит прогрессирующий характер. Со временем слабеют мышцы груди, шеи, исчезают сухожильные рефлексы, отмечаются нарушения глотания, слабый голос. Больного можно узнать по свисающей головке.

Патологию Кугельберга-Веландера диагностируют часто после 2 лет. Она считается относительно легкой формой СМА, многие больные доживают до 30-40 лет. Человек стоит, однако дается ему это с трудом из-за очень слабых мышц. Происходит постепенная атрофия мышц.
Ребенок до 10-12 лет развивается нормально, потом начинает спотыкаться, падает, теряет способность заниматься спортом, бегать, выходить из дома, просто перемещаться без инвалидного кресла. Больного мучают периодические судороги конечностей. Развивается сильный сколиоз, изменяется форма грудной клетки.
Часто у таких пациентов происходят переломы, отмечается ограниченный объем движения суставов.
К четвертому типу относят бульбоспинальную амиотрофию Кеннеди, дистальную амиотрофию Дюшенна-Арана, а также перонеальную амиотрофию Вюльпиана. Заболевания обычно диагностируются в возрасте 35-40 лет, иногда возрастные границы расширяются от 16 до 60 лет. Больной отмечает постепенную потерю мышечной силы, угасание рефлексов сухожилий, видимые сокращения мышц.
При атрофии Дюшенна-Арана прежде всего поражаются кисти рук. Амиотрофию Вюльпиана можно узнать по формированию крыловидных лопаток.
Причины и механизм развития заболевания
Спинальная амиотрофия развивается из-за мутировавшего SMN гена пятой хромосомы. Если оба родителя – его носители, существует 25%-ная вероятность, что ребенок родится больным.
Мутация гена SMN приводит к нарушению синтеза белка, в результате чего происходит разрушение мотонейронов спинного мозга. Нервные импульсы не проходят к мышцам, которые из-за бездействия атрофируются, человек теряет способность двигаться.
Считается, что теряет работоспособность сначала глубоко расположенная мускульная ткань.
Диагностика
Наиболее точным методом определения спинально-мышечной атрофии у детей является анализ ДНК. Он проводится как у родившегося малыша, так и во время внутриутробного развития. Дополнительно проводятся следующие исследования:

Если у молодых людей, планирующих рождение ребенка, есть родственники с патологией СМА, им рекомендовано пройти генетическую экспертизу.
Лечение
Основная цель исследований, направленных на терапию спинальной мышечной амиотрофии, связана с повышением уровня белка SMN. В настоящее время лекарственные препараты проходят испытания, и официальная российская медицина их не использует.
Лечение сегодня включает лекарства, которые улучшают прохождение нервных импульсов. Назначаются ноотропные препараты, основная задача которых – улучшение работы головного мозга. Назначаются биологически активные добавки, способствующие улучшению обмена веществ. Показана витаминотерапия, в частности, прием витаминов группы Б.
Средства влияющие на нервно-мышечную проводимость:
- Альфа-липоевая кислота
- Ацетил Л-карнитин
- Альфа-глицерофосфохолин
Витамины и витаминные комплексы:
- Тиамин (B-1)
- Пиридоксин (B-6)
- B-комплекс

Важными методами лечения являются массаж, физиотерапия, нейромышечная стимуляция. Назначается ЛФК. Физические упражнения помогают поддержать силу, с другой стороны, выполнение их в обществе, походы в бассейн помогают социализироваться, общаться с другими людьми.
Больным СМА рекомендовано соблюдение диеты. Продукты питания – источник веществ, необходимых мышцам. Так, необходимые аминокислоты содержатся в зерновых, мясе, рыбе, грибах, орехах, кисломолочных продуктах. Рекомендованы блюда из овса и пшеницы, бурого риса.
Естественному поддержанию и росту мышц поможет шпинат, брокколи, сельдь, лук, грейпфрут, арбуз. Для повышения тестостерона мужчинам рекомендуют принимать укроп, пастернак, женьшень, петрушку.
Прогноз
То, как будет развиваться болезнь, сколько лет проживет ребенок, зависит от ее типа.
При атрофии типа один прогноз крайне неблагоприятен. Около 50% малышей не доживают и до двух лет. Не больше 10% детей с болезнью Верднига-Гоффмана могут дожить до пяти лет. Причиной гибели чаще всего становится воспаление легких, остановка дыхания, сердца.
Пациенты, которым диагностирована болезнь Дубовица, живут в среднем до 10, иногда 12 лет. Около 30% малышей умирают, не достигнув четырех лет.
При SMA III типа детская смертность встречается реже. У многих пациентов симптомы появляются в предподростковом-подростковом возрасте. Через несколько лет они перестают ходить. Далее, по нарастающей, отмечается атрофия мышц внутренних органов, в том числе дыхательных.
Считается, что заболевание IV типа не влияет на продолжительность жизни, тем не менее, оно ведет к инвалидизации.
Профилактика
Мер, направленных на профилактику и предотвращение развития СМА, не существует. Женщина, ожидающая рождения ребенка, может заподозрить проблему, обратив внимание на слабость шевелений плода. Проведенный ДНК-анализ может подтвердить или развеять подозрения. При необходимости проводится медицинская комиссия, которая может порекомендовать прерывание беременности. Врач обязательно рассказывает о заболевании, его течении и последствиях.
После диагностики заболевания у уже родившегося ребенка его окружают заботой и вниманием. Использование системы искусственной вентиляции легких, отсасывателей мокроты, специальных приспособлений для движения малыша, который может передвигаться, помогают улучшить качество жизни и помочь ребенку жить. Рекомендовано регулярно делать массаж, физиопроцедуры. Детей даже с ограниченными движениями возят в бассейн.
Спинальная амиотрофия – опасная, пока не поддающаяся лечению патология. Она характеризуется атрофией мышц. Возникает в разном возрасте. Прогноз в большинстве случаев неблагоприятный.
Для подготовки статьи использовались следующие источники:
Селиверстов Ю. А., Клюшников С. А., Иллариошкин С. Н. Спинальные мышечные атрофии: понятие, дифференциальная диагностика, перспективы лечения // Журнал Нервные болезни — 2015
Лепесова М. М., Ушакова Т. С., Мырзалиева Б. Д. Дифференциальная диагностика спинальной мышечной амиотрофии первого типа // Вестник Алматинского государственного института усовершенствования врачей — 2016
Читайте также:
