
Что такое спинальная амиотрофия кугельберга-веландера
Спинальная мышечная атрофия представляет собой редкую группу наследственных заболеваний, которые вызывают прогрессивную дегенерацию клеток переднего рога спинного мозга. Точная причина вырождения неизвестна. Потеря этих клеток приводит к прогрессирующей болезни двигательного нейрона. А вот спинальная мышечная атрофия Кугельберга-Веландера, является (одной из) мягкой формой спинальной мышечной атрофии, симптомы и проявления которой, как правило, начинают проявляться в возрасте после 18 месяцев.

Мальчик с атрофией спинальных мышц
Спинальная мышечная атрофия была впервые описана в 1890-х годах Гвидо Верднигом, врачом из университета Вены. Вскоре после этого, профессор Иоганн Гофман из Гейдельбергского университета представил документ, описывающий синдром прогрессирующей атрофии, слабости и смерти братьев и сестер в раннем периоде детства с генетически нормальными родителями. Оба врача провели вскрытие своих пациентов и обнаружили серьезные атрофии брюшных корешков спинного мозга. Они также обнаружили, гистологические признаки потери двигательных нейронов в клетках передних рогов этого региона. Гофман назвал этот синдром как spinale muskelatrophie (спинальная мышечная атрофия).
В начале 1960-х годов, Байерс разделил все атрофии на категории по степени тяжести и по возрасту, в котором начинается развитие симптомов, в целях прогнозирования исхода для пациента. Его система, она приведена ниже, стала основой для разработки наиболее широко признанной системы, которая в настоящее время используется в классификации всех спинальных мышечных атрофий.
- Симптомы начинаются в возрасте до 6 месяцев
- Этот тип также известен как спинальная мышечная атрофия с инфантильным началом, или болезнь Верднига-Гоффмана
- Симптомы начинают развиваться в возрасте 6-18 месяцев
- Этот тип также известен как хроническая спинальная мышечная атрофия или промежуточная спинальная мышечная атрофия.
- Симптомы начинают развиваться в возрасте от 18 месяцев, как правило, в конце детского или уже в подростковом возрасте
- Этот тип также известен как синдром Кугельберга-Веландера или мягкая спинальная мышечная атрофия.
- Эта категория предназначена для тех атрофий, симптомы которых начинают проявляться только в начале взрослой жизни.
- Это расстройство обычно имеет гораздо более благоприятный прогноз, чем другие типы.
В этой статье мы будет фокусироваться только на типах III и IV.
Спинальная мышечная атрофия Кугельберга-Веландера. Причины
Точная этиология спинальной мышечной атрофии Кугельберга-Веландера неизвестна. Спинальные мышечные атрофии являются наследственными заболеваниями, которые почти всегда имеют аутосомно-рецессивный признак. Все формы спинальной мышечной атрофии связаны с делециями генов на длинном плече хромосомы 5, в полосе 5q13. Эти гены включают в себя SMN1 и SMN2. А ген SMN1, как полагают, является первичным болезнетворным геном. Белок SMN1 связан со сборкой специальных рибонуклеопротеинов, которые имеют решающее значение в обработке мРНК. Белок SMN1 также помогает регулировать запрограммированную гибель клеток (апоптоз). Некоторые исследователи предположили, что делеции в генах SMN могут быть связаны с нарушениями в метаболизме 3′,5′-аденозинмонофосфата. Но способствуют ли эти нарушения дегенерации нейронов еще не известно и это еще предстоит увидеть.
Спинальная мышечная атрофия Кугельберга-Веландера. Патофизиология
Спинальная мышечная атрофия развивается из-за последовательной дегенерацией клеток и структур двигательных мышц. Мышечная атрофия, вызванная прогрессирующей потерей передних рогов в спинном мозге, является универсальной. Также могут быть вовлечены двигательные нервы в нижней части мозга, как правило, это касается черепных нервов V-XII (V, VII, IX, XII). В этих местах можно гистологически наблюдать различные этапы дегенерации. По мере уменьшения количества нервных клеток, исследователи могут отметить глиоз, пикноз и валлеровскую дегенерацию в периферических нервах. Эти процессы обычно начинаются на каудальном конце и они, как правило, являются симметричными. Нижние конечности обычно поражаются раньше и глубже, чем верхние. Это вырождение наиболее часто влияет на ближнюю мускулатуру.
Спинальная мышечная атрофия Кугельберга-Веландера. Симптомы и проявления
- Пациенты с спинальной мышечной атрофией Кугельберга-Веландера отмечают коварное начало развития слабости, которая часто развивается после короткого периода болезни, например, после гриппа.
- Пациенты чаще сообщают о симптомах, связанных со слабостью разгибательных, тазобедренных и суставных мышц. Слабость в этих мышцах часто описывается трудностями при подъеме по лестнице или при подъеме тела из положения сидя на полу.
- Некоторые пациенты также могут ощущать мягкий тремор и иногда болезненные мышечные спазмы.
- Сложность в ходьбе или в беге.
- Родители детей, с спинальной мышечной атрофией Кугельберга-Веландера, могут сообщить о задержках в развитии или о снижении спортивных способностей у своих детей.
- Проксимальная мышечная слабость, в большей своей степени, проявляется в тазовом поясе, а не в плечевом.
- Пациенты могут проявлять снижение мышечного тонуса, уменьшение глубоких сухожильных рефлексов.
- Можно обнаружить легкий тремор, если пациента попросят выпрямить пальцы. Это является результатом денервации с последующей реиннервацией и асинхронной стрельбой реструктурированных и увеличенных моторных клеток.
- За пациентами можно обратить внимание на переваливающуюся походку.
- Примерно одна треть больных имеют слабость жевательных мышц.
- Сенсорные данные в норме.
Спинальная мышечная атрофия Кугельберга-Веландера. Диагностика
- Молекулярно-генетическое тестирование. Текущее диагностическое тестирование спинальной мышечной атрофии Кугельберга-Веландера включает в себя адресный анализ мутаций в экзонах 7 и 8 гена SMN1.
- Другие тесты - уровни креатин киназы могут быть подняты.
- УЗИ мышц может быть проведено для оценки нейрогенной атрофии при спинальной мышечной атрофии Кугельберга-Веландера, но это довольно неспецифично. И уже сегодня, УЗИ потеряло свои преимущества в качестве диагностического инструмента в диагностике этой атрофии. Сканирование мозга не показывает никаких отклонений в головном мозге.
- Анализ тканей полученных на биопсии мышц могут указать на свидетельства нейрогенной атрофии и хронической реиннервации. Каркасные изменения, при спинальной мышечной атрофии Кугельберга-Веландера, включают в себя сочетание узких, больших, гипертрофированных волокон. Эти волокна отделяются друг от друга обильными жировыми и волокнистыми тканями.
- Электромиография (ЭМГ) и исследования нервной проводимости могут быть очень полезны для врача в диагностике спинальной мышечной атрофии Кугельберга-Веландера. Диффузные изменения на ЭМГ наблюдаются в конечностях и в бульбарной мускулатуре. Полученные данные согласуются с дегенерацией аксонов.
Спинальная мышечная атрофия Кугельберга-Веландера. Лечение
Сегодня не известно никакого известного лечения спинальной мышечной атрофии Кугельберга-Веландера. Таким образом, уход за пациентом будет ориентирован на симптоматический контроль и на профилактическое оздоровление. Поддержание подвижности суставов пациента очень важно, потому что цель будет заключаться в снижении тяжести контрактур. Растяжка и силовая тренировка у пациентов под присмотром опытного физиотерапевта – очень важный компонент профилактической реабилитации. Для пациентов школьного возраста, физиотерапевт может провести консультацию относительно соответствующих адаптивных подходов. Водная терапия также является отличным способом поддержания мобильности, прочности и гибкости суставов.
Из-за прогрессирующей слабости, связанной с спинальной мышечной атрофией Кугельберга-Веландера, пациентам может потребоваться постоянное использование инвалидной коляски.
Трудотерапия также играет важную роль в решении индивидуальных потребностей пациентов. Трудотерапия является полезной в повышении независимости в повседневной жизни пациента.
Если у пациента будет развиваться сколиоз, то некоторым из них может потребоваться хирургическое вмешательство. Сухожильные удлинения могут быть необходимы для улучшения функциональности суставов.
Спинальная мышечная атрофия Кугельберга-Веландера. Осложнения
- Ортопедические осложнения. Сколиоз является серьезной проблемой у половины пациентов со спинальной мышечной атрофией Кугельберга-Веландера. Эти пациенты должны регулярно проходить рентгенографию, и некоторым из них, возможно, потребуется операция или ортез, который может помочь в сдерживании деформации позвоночника.
- Подвывих сустава является обычным явлением.
- Дыхательные проблемы – легочные заболевания являются основной причиной тяжелой заболеваемости и высокой смертности у пациентов с атрофией. Дисбаланс в силе выдоха и вдоха, приводит к нарушениям в дыхании, гиповентиляции во время сна и к повышенному потенциалу развития рецидивирующих инфекций. Один из исследователей опубликовал в докладе данные, согласно которым, у пациентов в возрасте 17 лет, легочная функция сократилась до 79%.
- Нарушения сна. Пациенты могут указывать на увеличение дневной усталости от ночного храпа и апноэ.
- Контрактуры.
- Дисфагия
Спинальная мышечная атрофия Кугельберга-Веландера. Прогноз
У пациентов с спинальной мышечной атрофией, это состояние будет прогрессировать до потери двигательной функции.
Спинальная мышечная атрофия типа III и IV, в отличие от типов I и II, согласуются с нормальной продолжительностью жизни. Большинство пациентов будут использовать инвалидную коляску уже с четвертого десятилетия жизни.
Амиотрофия Кугельберга-Веландера — это спинальная мышечная атрофия, отличающаяся поздним развитием и наиболее доброкачественным течением. Характерны атрофии мышц тазового пояса и бедер с последующим присоединением атрофий плечевого пояса и плеч, сочетание мышечной слабости и гипотонии с фасцикулярными подергиваниями, наличие псевдогипертрофий. Диагностика проводится при помощи ЭФИ нервной системы, исследования мышечного биоптата, ДНК-анализа, МРТ позвоночника. Лечение симптоматическое и малоэффективное, но медленное прогрессирование симптомов обуславливает длительную двигательную способность пациентов.
МКБ-10

- Причины
- Патогенез
- Симптомы
- Осложнения
- Диагностика
- Дифференциальная диагностика
- Лечение амиотрофии Кугельберга-Веландера
- Консервативная терапия
- Экспериментальное лечение
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Амиотрофия Кугельберга-Веландера — относительно доброкачественный тип спинальной амиотрофии с медленным прогрессированием и поражением преимущественно мышечных групп проксимальных отделов конечностей. Подробно описана в 1956 г. швейцарскими врачами Е. Кугельбергом и Л. Валандером, в честь которых и была названа. В практической неврологии и генетике употребляются также другие названия: ювенильная амиотрофия, спинальная мышечная атрофия (СМА) III типа.
Амиотрофия Кугельберга-Веландера клинически манифестирует после 2-летнего возраста, наиболее часто в период с 3 до 10 лет. Известны отдельные случаи более позднего дебюта в возрасте 14-30 лет. Точные данные о распространенности СМА III типа пока отсутствуют. Некоторые авторы до сих пор считают данный тип амиотрофии поздней формой болезни Верднига-Гоффмана.
Причины
Патогенез
Нарушения генетического аппарата обуславливают появление и прогрессирование дегенеративных изменений в передних рогах спинного мозга. Процесс затрагивает локализующиеся там двигательные нейроны (мотонейроны). Характерно первоначальное поражение нижнегрудных и поясничных спинальных сегментов, приводящее к мышечной слабости в проксимальных отделах ног, а затем поражение верхнегрудных и шейных сегментов со слабостью в мышцах плечевого пояса.
Симптомы
От других типов СМА форма Кугельберга-Веландера отличается наличием фасцикулярных подергиваний в пораженных мышцах, а иногда и в мышечных группах голеней и предплечий. Зачастую отмечаются тремор языка, мелкоразмашистый тремор пальцев.
Осложнения
В отличие от других разновидностей спинальных мышечных атрофий (I и II типа), амиотрофия Кугельберга-Веландера характеризуется более доброкачественным течением. Возможно формирование костных деформаций: искривления позвоночника, деформации грудной клетки и стоп, в отдельных случаях ‒ сухожильных ретракций и контрактур суставов. Однако костно-суставные проявления носят умеренный характер.
Глубокая инвалидизация с утратой способности к ходьбе и самостоятельному обслуживанию наступает крайне редко и только на поздних стадиях заболевания. К самым распространенным осложнениям можно отнести нарушения равновесия и походки вследствие слабости мышц тазового пояса и проксимальных отделов нижних конечностей. Частые падения повышают риск патологических переломов.
Диагностика
Больных с амиотрофией Кугельберга-Веландера курируют врачи-педиатры и детские неврологи. Важны анамнестические данные: возраст дебюта заболевания (после 2-х лет), характер прогрессирования, наличие близкого родственника с этой патологией. При осмотре обращается внимание на снижение тонуса мышц нижних конечностей, ослабление или утрату коленного и ахиллова рефлексов, скелетно-мышечные деформации (искривление позвоночника, контрактуры суставов).
Также у 50% пациентов отмечается типичная псевдогипертрофия икроножных мышц, т.е. их увеличение за счет отложения жира и разрастания соединительной ткани. Чтобы подтвердить или исключить диагноз назначается следующее дополнительное обследование:
Дифференциальный диагноз амиотрофии Кугельберга-Веландера следует проводить с другими нейродегенеративными заболеваниями, манифестирующими в детском возрасте. К ним можно отнести:
- детский церебральный паралич;
- псевдогипертрофическую мышечную дистрофию Дюшенна;
- конечностно-поясную форму прогрессирующей мышечной дистрофии.
Лечение амиотрофии Кугельберга-Веландера
Для прохождения лечения пациентов необходимо госпитализировать в стационар. На сегодняшний день не существует этиотропной терапии с доказанной эффективностью. Все мероприятия носят симптоматический и паллиативный характер. Тем не менее, комплексный подход и четкое соблюдение рекомендаций специалистов позволяют значительно улучшить качество жизни больного. Применяются следующие виды лечения:
- Физиотерапия. Для улучшения микроциркуляции в мышечной ткани проводятся сеансы электростимуляции модулированным током, электрофорез, грязевые аппликации.
- ЛФК. С целью замедления атрофии мышц необходимо регулярное выполнение различных физических упражнений. Предпочтение отдается занятиям, направленным на укрепление мышц нижних конечностей – приседание, бег и пр.
- Ортопедия. Для коррекции и предупреждения скелетно-мышечных деформаций и суставных контрактур используются ортопедические приспособления (ортезы, корсеты и т.д.).
- Медикаменты. Для улучшения обменных процессов в мотонейронах из лекарственных средств применяются метаболические препараты (коэнзим Q10, L-карнитин), витамины группы В (В1, В6, В12) в высоких дозах.
Ведутся многочисленные исследования по поиску и разработке лекарственных препаратов, влияющих на ключевые звенья патогенеза амиотрофии Кугельберга-Веландера, что позволит значительно замедлить или полностью остановить прогрессирование нейродегенеративных процессов. Завершилась II фаза клинических испытаний препарата Рисдиплам (RG7916) – модификатора сплайсинга мРНК гена SMN. Препарат показал высокую терапевтическую эффективность при всех типах СМА.
Прогноз и профилактика
Благодаря своему медленному течению и позднему возникновению, амиотрофия имеет относительно благоприятный прогноз. Длительное время пациенты сохраняют способность самостоятельно двигаться, а в случаях более позднего начала (в 20-30 лет) доживают до старости, не теряя способности к самообслуживанию.
Единственный способ предотвратить развитие заболевания – пренатальная диагностика с последующим прерыванием беременности. В биоптатах ворсин хориона или амниотической жидкости обнаруживается мутация SMN. Вторичная профилактика заключается в предупреждении возникновения неблагоприятных последствий (костно-суставных деформаций).
Данное заболевание отличается поздним проявлением на фоне доброкачественного течения с медленным прогрессированием поражения мышечных тканевых соединений проксимальных отделов конечностей. Амиотрофия развивается в возрасте от 3 до 10 лет. В некоторых случаях заболевание может дебютировать и в более позднем возрасте – от 14 до 30 лет.
Амиотрофия спинальная доброкачественная Кугельберга-Веландера, как и все другие формы спинальной амиотрофии, передается наследственным путем по аутосомно-рецессивному типу. Данный тип характеризуется наличием мутационного гена как у матери, так и у отца.
Генетические нарушения обуславливают развитие изменений дегенеративной формы в передних отделах спинного мозга. Патологический процесс также затрагивает мотонейроны, характерным является первоначальное поражение спинальных сегментов нижнегрудной и поясничной области, в результате чего появляется слабость в мышечных соединениях проксимального отдела нижних конечностей. Позже поражаются верхнегрудная и шейная область, развивается слабость в мышечной системе плечевого пояса.
Заболевание начинает проявляться в период, когда малыш уже может самостоятельно передвигаться. Первым симптомом является повышенная утомляемость после долгой ходьбы или в процессе бега. Большая часть пациентов отмечает характерную неустойчивость и неуклюжесть, происходят частые падения, могут возникать неудобства при поднятии по лестнице. Таким образом, со временем происходит атрофия мышечных тканей бедер и тазовой области.
Через несколько лет появляются такие признаки:
- поражения проксимальных мышечных групп рук;
- снижение активности в плечевом поясе и верхних конечностях;
- атрофические процессы в мышечных соединениях плечевого пояса и области лопаток, формируется „крыловидность” лопаток;
- симметричность мышечных атрофий;
- нарушение мышечной силы в мышцах, которые поражены;
- угасание рефлексов сухожилий в плечевой области;
- нарушение рефлексов коленных сухожилий;
- фасцикулярные подергивания в мышечных тканях;
- тремор языка;
- тремор пальцев рук мелкоразмашистой формы;
- в тяжелых случаях наблюдается деформация позвоночника и грудной клетки, стоп.
Данное заболевание исследует невролог и генетик. При этом учитываются такие факторы:
- возраст начальной стадии;
- скорость проявления и прогрессирования симптоматики;
- результаты генетического исследования.
Главными критериями, по которым устанавливается правильный диагноз, являются:
- развитие патологических процессов в возрасте после 2 лет;
- аутосомно-рецессивный тип;
- поражение нижних и верхних конечностей;
- псевдогипертрофия;
- наличие тремора и фасцикуляций.
Дополнительно рекомендуется уточнение диагноза с использованием диагностирования ДНК. Также необходимо провести биохимию крови, которая отмечается при данном заболевании повышенным уровнем креатинфосфокиназы.
Электронейромиография даст возможность установить характер поражений и предотвратить развитие миопатии, а биопсия мышц – определить пучковые атрофические процессы в мышечных волокнах. Магнитно-резонансная томография позвоночника может быть использована как дополнительная диагностика для определения органических патологий:
- новообразования в спинном мозге;
- миелит;
- гематомиелия и другие.
На сегодняшний день еще не разработаны патогенетические методы лечения. Терапевтический курс направлен на улучшение метаболических процессов в нервных тканевых соединениях и работы пораженных нервно-мышечных структурных компонентов.
В качестве лечения назначается прием витаминов группы В, L-карнитин, также рекомендуется лечебная физкультура, которая даст возможность замедлить сокращение рефлексов опорно-двигательного аппарата. Взрослые пациенты должны правильно трудоустроиться, так как работа должна быть без избыточных мышечных напряжений.
Амиотрофия Кугельберга-Веландера имеет хороший прогноз, так как долгое время сохраняется способность передвигаться без посторонней помощи, а при более позднем возникновении люди с данным диагнозом доживают до преклонного возраста и могут самостоятельно себя обслуживать.
- Аллергические заболевания
- Женские болезни
- Кожные заболевания
- Болезни желудочно-кишечного тракта
- Сексуальные расстройства
- Мужские болезни
- Заболевания опорно-двигательной системы
- Неврологические болезни
- ЛОР-болезни
- Проктологические заболевания
- Болезни волос и кожи головы
- Болезни глаз
- Болезни зубов и полости рта
- Болезни молочных желез
- Болезни органов дыхания
- Болезни сердца и сосудов
- Болезни суставов
- Венерические заболевания
- Детские болезни
- Заболевания головного мозга
- Инфекционные заболевания
- Наркологические болезни
- Неотложные состояния
- Общие заболевания
- Онкологические заболевания
- Психические болезни
- Урологические болезни
- Хирургические болезни
- Эндокринологические болезни
Всегда самая актуальная, полная и полезная информация о медицинских учреждениях, лабораториях, медцентрах и клиниках Молдовы и других стран.
Здесь вы найдете экспертные статьи о заболеваниях, методах лечения, способах профилактики, интервью с практикующими врачами и другими специалистами, а также новости от наших партнеров.
Адрес: Республика Молдова, г. Кишинев, ул. Влайку Пыркэлаб, 30/1
Отдел рекламы: br> Телефоны: +373 68 199 951; +373 68 585 053
Хроническая спинальная мышечная атрофия III типа (СМА третьего типа, болезнь Вольфарта-Кугельберга-Веландер, псевдомиопатическая СМА, болезнь Кугельберга, болезнь Кугельберга-Веландер) также передается по аутосомно-рецес-сивному типу, хотя нередки и спорадические случаи. Она также в большинстве случаев связана с делецией в гене SMN1. Заболевание может начаться в любой момент грудного возраста или раннего детства и является чрезвычайно коварным.
Часто различают два подтипа: пациенты с подтипом IIIa начинают ходить в возрасте до трех лет, а пациенты с подтипом IIIb — после этого возраста, и данное различие может иметь отношение к прогнозу (Rudnik-Schoneborn et al., 2001).
Болезнь прогрессирует очень медленно в направлении дистальных отделов нижних конечностей и/или проксимальных отделов верхних конечностей. Часто встречается полая стопа. Многие пациенты демонстрируют грубый тремор кистей (Dawood и Moosa, 1983), который, вероятно, связан с несовершенной синхронизацией уменьшенного числа больших моторных единиц в результате коллатеральной реиннервации мышечных волокон оставшимися нейронами. ЭКГ показывает дрожание изолинии, по-видимому, отражающее тот же самый мышечный тремор.
Наиболее пострадавшие люди могут продолжить вести нормальную жизнь во взрослом возрасте (Zerres et al., 1995, 1997), но некоторые случаи протекают быстрее; ортопедические осложнения могут произвести к тяжелым функциональным ограничениям, которых желательно избегать в ходе адекватной физиотерапии и/или ортопедического лечения. Правильное консультирование с настоятельной рекомендацией приобретения навыков, позволяющих самостоятельное проживать длительное время, является крайне важным. В редких случаях прослеживается связь с врожденной (Oka et al., 1995) или прогрессирующей (Alberfeld и Namba, 1969) офтальмоплегией.
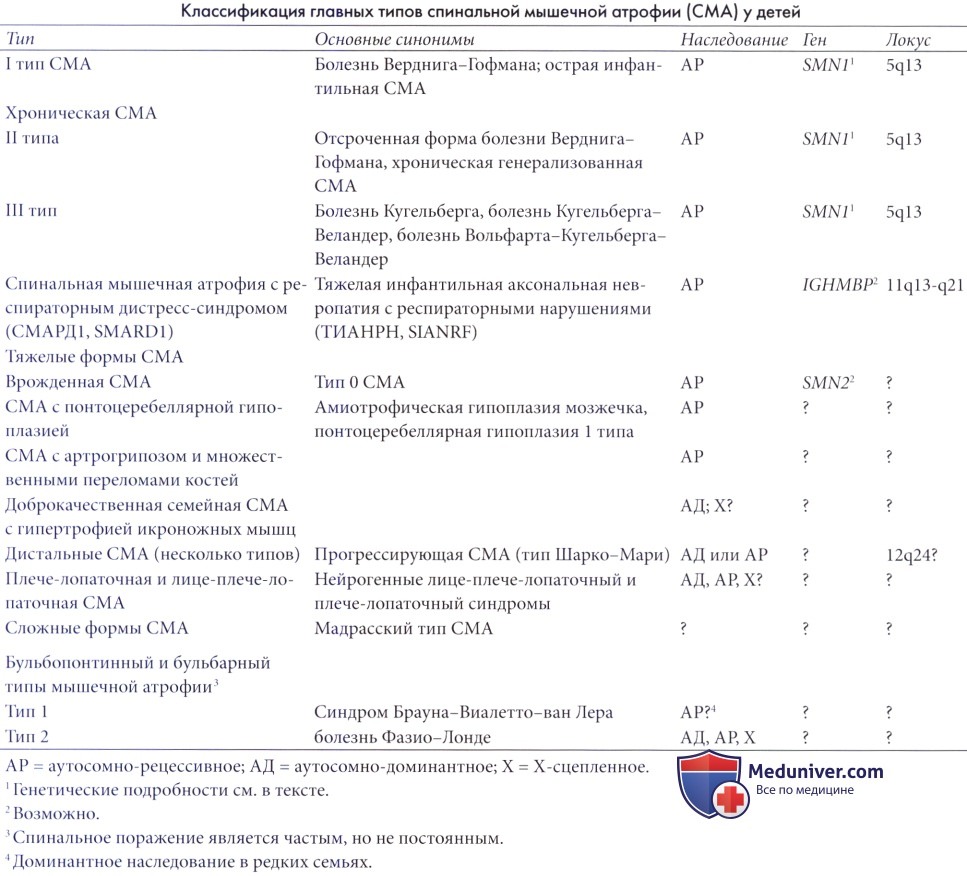
Лечение СМА III типа основано на физиотерапии и профилактике контрактур и ретракций. Для обоих типов проведено несколько испытаний препаратов, способных вызвать изменения в чтении ДНК. Они включают габапентин (Miller et al., 2001; Merlini et al., 2003), рилузол (Russman et al., 2003), фенилбутират (Mercuri et al., 2004a). Несмотря на некоторые положительные результаты, ни один из них не доведен до стадии практического применения. Генная терапия находится в стадии изучения (DiDonato et al., 2003).
Молекулярно-генетический диагноз рецессивных проксимальных СМА. Диагностика трех классических типов СМА методами молекулярной генетики возможна главным образом за счет демонстрации гомозиготной делеции 7 экзона в гене SMN1 (Melki et al., 1994). Однако делеция отсутствует в меняющейся, но немногочисленной группе пациентов с СМА; редкие люди с гомозиготной делецией могут иметь нормальный фенотип; и отношение генов SMN к СМА еще не полностью определено.
Приблизительно 10% случаев СМА (например, СМАРД1, СМА с оливопонтоцеребеллярной атрофией), не связаны с делецией или связаны с 5 хромосомой (Novelli et al., 1995; Rudnick-Schoneborn et al., 1995). Напротив, ряд случаев с наличием одного или нескольких принятых критериев исключения (Munsat и Davies, 1992) может демонстрировать типичную делению, включая случаи с дисфункцией центральной нервной системы, артрогрипозом или сочетанными пороками развития (Rudnik-Schoneborn et al., 1996).
Анализ ДНК обеспечивает возможность достоверного пренатального диагноза большинства типов классической СМА (Cobben et al., 1993; Wirth et al., 1995). Можно провести тест ДНК с использованием ПЦР (Van der Steege et al., 1995). Суть теста заключается в демонстрации гомозиготной делеции 7 экзона гена SMN. Он относительно легко выполним, но может быть негативным приблизительно у 2% пораженных детей, которые являются носителями отличающегося дефекта ДНК (Raymond, 1997).
Редактор: Искандер Милевски. Дата публикации: 10.1.2019
Спинальная амиотрофия Верднига–Гоффманна. Заболевание описано Дж. Верднигом в 1891 г. и Ж. Гоффманном в 1893 г. Частота 1 на 100 000 населения, 7 на 100 000 новорожденных. Наследуется по аутосомно‑рецессивному типу.
Клинические проявления. Выделяют три формы заболевания: врожденную, раннюю детскую и позднюю, различающиеся временем проявления первых клинических симптомов и темпом течения амиотрофического процесса.
Течение. Болезнь имеет быстро прогрессирующее течение. Летальный исход наступает до 9‑летнего возраста. Одной из основных причин смерти являются тяжелые соматические расстройства (сердечно‑сосудистая и дыхательная недостаточность), обусловленные слабостью мускулатуры грудной клетки и снижением участия ее в физиологии дыхания.
При ранней детской форме первые признаки болезни возникают, как правило, на втором полугодии жизни. Моторное развитие в течение первых месяцев удовлетворительное. Дети своевременно начинают держать голову, сидеть, иногда стоять. Заболевание развивается подостро, нередко после инфекции, пищевой интоксикации. Вялые парезы первоначально локализуются в ногах, затем быстро распространяются на мышцы туловища и руки. Диффузные мышечные атрофии сочетаются с фасцикуляциями, фибрилляциями языка, мелким тремором пальцев, сухожильными контрактурами. Мышечный тонус, сухожильные и надкостничные рефлексы снижаются. В поздних стадиях возникают генерализованная мышечная гипотония, явления бульбарного паралича.
Течение. Злокачественное, хотя и мягче по сравнению с врожденной формой. Летальный исход наступает к 14–15 годам жизни.
Течение. Злокачественное, но мягче, чем у первых двух форм. Нарушение способности самостоятельной ходьбы происходит в 10–12‑летнем возрасте. Больные живут до 20–30 лет.
Диагностика и дифференциальный диагноз. Диагноз строится на основании данных генеалогического анализа (аутосомно‑рецессивный тип наследования), особенностей клиники (раннее начало, наличие диффузных атрофии с преимущественной локализацией в проксимальных группах мышц, генерализованной мышечной гипотонии, фасцикуляций и фибрилляций языка, отсутствие псевдогипертрофий, прогредиентное и в большинстве случаев злокачественное течение и др.), результатах глобальной (накожной) и игольчатой электромиографии и морфологического исследования скелетных мышц, позволяющего выявить денервационный характер изменений.
Лечение. При спинальной амиотрофии Верднига–Гофмана назначают ЛФК, массаж, препараты, улучшающие трофику нервной ткани – церебролизин, аминалон (гаммалон), пиридитол (энцефабол).
Спинальная юношеская псевдомиопатическая мышечная атрофия Кугельберга–Веландера. Частота не установлена. Наследуется по аутосомно‑рецессивному, реже – по аутосомно‑доминантному, рецессивному сцепленному с Х‑хромосомой типу.
Патоморфология. Обнаруживаются недоразвитие и дегенерация клеток передних рогов спинного мозга, демиелинизация передних корешков, дегенерация двигательных ядер IX, X, XII черепных нервов. В скелетных мышцах – сочетанные изменения, типичные для нейрогенных амиотрофии (пучковая атрофия мышечных волокон) и первичных миодистрофий (атрофии и гипертрофии мышечных волокон, гиперплазия соединительной ткани).
Клинические проявления. Первые признаки заболевания проявляются в 4–8 лет. Описаны случаи начала болезни и в более позднем возрасте – 15–30 лет. В начале болезни характерными симптомами являются патологическая мышечная утомляемость в ногах при длительной физической нагрузке (ходьба, бег), иногда спонтанные подергивания мышц.
Течение. Болезнь медленно прогрессирует.
Диагностика и дифференциальный диагноз. Диагноз ставится на основании данных генеалогического анализа (аутосомно‑рецессивный, аутосомно‑доминантный, рецессивный сцепленный с Х‑хромосомой тип наследования), особенностей клиники (начало болезни преимущественно в возрасте 4–8 лет, симметричные атрофии мышц, распространяющиеся по восходящему типу фасцикуляции мышц, мелкий тремор языка, псевдогипертрофии икроножных мышц, медленое прогредиентное течение), результатов глобальной и игольчатой электромиографии и морфологического исследования скелетных мышц, позволяющего выявить денервационный характер изменений.
Дифференцировать болезнь следует от прогрессирующих мышечных дистрофий Беккера, Эрба–Рота, спинальной амиотрофии Верднига–Гоффманна.
Наследственная дистальная спинальиая амиотрофия. Частота не установлена. Наследуется по аутосомно‑рецессивному, реже – по аутосомно‑доминантному, рецессивному сцепленному с Х‑хромосомой типу.
Патоморфология. Соответствует другим спинальным амиотрофия м.
Клинические проявления. Первые признаки заболевания проявляются преимущественно в первой декаде жизни. Начальными симптомами болезни являются слабость и атрофия дистальной мускулатуры ног. В 25 % случаев наблюдаются слабость и атрофия дистальной мускулатуры рук. Отличительные особенности – грубые деформации стоп, ранняя утрата ахиллова рефлекса при сохранности коленных и глубоких рефлексов с рук, отсутствие чувствительных расстройств.
Течение. Болезнь медленно прогрессирует.
Диагностика и дифференциальный диагноз. Диагноз ставится на основании генеалогического анализа (аутосомно‑доми‑нантный, аутосомно‑рецессивный, рецессивный сцепленный с Х‑хромосомой тип наследования), особенностей клинической картины (начало в первой декаде жизни, преимущественная локализация атрофии в дистальных отделах нижних конечностей, грубые деформации стоп, отсутствие чувствительных нарушений, медленное прогрессирование миодистрофического процесса), результатов глобальной и игольчатой электромиографии, позволяющей выявить вовлечение в процесс передних рогов спинного мозга.
Дифференцировать заболевание следует от дистальной миопатии Говерса–Веландера, невральной амиотрофии Шарко–Мари–Тута.
64. Аутосомно-доминантная миотония (болезнь Томсена). Клиника, диагностика, лечение.
Миотонии – гетерогенная группа нервно‑мышечных заболеваний, объединенная общим характерным комплексом нарушений мышечного тонуса, проявляющимся затруднением расслабления мышц после активного сокращения.
Различают наследственные миотонии (стационарные медленно прогрессирующие и периодические, рецидивирующие формы) и миотонические синдромы.
Врожденная миотония (болезнь Лейдена–Томсена). Заболевание впервые описано Лейденом в 1874 г. Томсен в 1876 г. обратил внимание на наследственную природу болезни на примере своей семьи (дети и многие родственники – 20 членов его семьи в 4 поколениях страдали миотонией).
Частота 0,3–0,7 на 100 000 населения. Наследуется по аутосомно‑доминантному типу. Пенетрантность более высокая у лиц мужского пола.
Патогенез. Имеют значение нарушения проницаемости клеточной мембраны, изменение ионного и медиаторного обмена (нарушения функциональной взаимосвязи в звене кальций–тропонин–актомиозин), повышенная чувствительность ткани к ацетилхолину и калию.
Патоморфология. При световой микроскопии обнаруживается гипертрофия отдельных мышечных волокон; гистохимически определяется уменьшение размеров II типа мышечных волокон; при электронной микроскопии выявляются умеренная гипертрофия саркоплазматической сети, изменение формы и увеличение размера митохондрий, расширение телофрагмы миофибриллярных волокон.
Течение. Болезнь медленно прогрессирует. Трудоспособность сохраняется в течение длительного времени.
Диагностика и дифференциальный диагноз. Диагноз строится на основании генеалогического анализа (аутосомно‑доминантный тип наследования), особенностей клинической картины (атлетический тип телосложения, диффузные гипертрофии мышц, миотонический синдром), данных глобальной электромиографии (миотоническая реакция).
Дифференцировать заболевание следует от других форм миотоний, иногда – от псевдогипертрофических форм прогрессирующих мышечных дистрофий.
Лечение. Назначают дифенин (по 0,1–0,2 г 3 раза в день в течение 2–3 нед), диакарб (по 0,125 г 2 раза в день в течение 2–3 нед), препараты кальция (внутривенно 10 % раствор хлорида кальция по 10 мл или глюконат кальция внутримышечно). Предполагается, что дифенин оказывает тормозящее влияние на моно– и полисинаптическое проведение в ЦНС, а диакарб изменяет проницаемость мембран. Целесообразны физиотерапия в виде гальванического воротника и трусов с кальцием, лечебная гимнастика.
65. Дистрофмческая миотония (болезнь Штейнерта-Куршмана). Клиника, диагностика, лечение.
Дистрофическая миотония Россолимо–Штейнерта–Куршмана. Заболевание впервые описано Г. И. Россолимо в 1901 г., а впоследствии Штейнертом и Куршманом в 1912 г. Частота 2,5–5 на 100 000 населения. Наследуется по аутосомно‑доминантному типу.
Патогенез. Неясен. Предполагается первичный дефект мембран.
Патоморфология. Методом световой микроскопии обнаруживают сочетание атрофированных и гипертрофированных мышечных волокон, разрастание соединительной ткани, замещение мышечной ткани жировой и соединительной. При электронной микроскопии определяются изменение размеров митохондрий, деструкции миофибриллярного аппарата, саркоплазматической сети.
Заболевание медленно прогрессирует.
Диагностика и дифференциальный диагноз. Диагноз ставится на основании данных генеалогического анализа (аутосомно‑доминантный тип наследования), особенностей клиники (сочетание миотонических, миопатических, нейроэндокринных, сердечно‑сосудистых нарушений), результатов глобальной электромиографии (миотоническая реакция), биохимического исследования крови (инсулинорезистентность).
Дифференцировать заболевание следует от врожденной миотонии Томсена, других миотонических форм, прогрессирующих мышечных дистрофий – дистальной миопатии, невральной амиотрофии.
Лечение. Как и при врожденной миотонии, положительный эффект дают дифенин, диакарб. Показано применение анаболических стероидов (ретаболил, неробол, метиландростендиол). В диете следует уменьшить содержание калия.
Читайте также:
