
Симптом запястья при синдроме марфана
Болезнь Марфана, или синдром Марфана, относится к числу наследственных болезней соединительной ткани. Клиническое описание этого заболевания было дано впервые фран-цузским педиатром А. Марфаном (1896). Ашар (1902) назвал его арахнодактилией и долихосте-номелией. Второе наиболее известное название — синдром Марфана — предложено Бергером (1915).
В этиологии и патогенезе синдрома Марфана большое значение придают поражению со-единительной ткани, а именно нарушению синтеза коллагена, которое приводит к накоплению фракций растворимого (незрелого) коллагена с распадом его на метаболиты, содержащие окси-пролин.
Заболевание проявляется уже в детском возрасте, но большинство пациентов — молодые люди. Нередко болезнь приводит к ранней инвалидизации и ранней смерти; встречается с рав-ной частотой у мужчин и женщин.
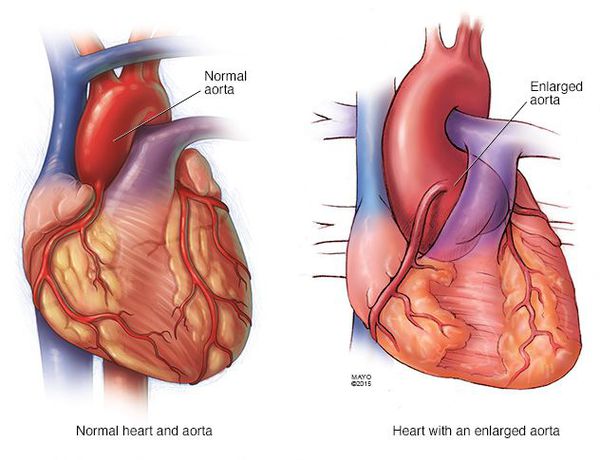
Типичным для синдрома Марфана считают сочетание характерных изменений опорно-двигательного аппарата (долихостеномелия, арахнодактилия), глаз (подвывих хрусталика) и сердечнососудистой системы (эрдхеймовский некроз, аневризма аорты).
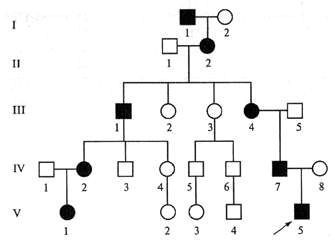
Доминантный характер наследования предполагает как один из механизмов естественно-го отбора наличие стертых форм заболевания, диагностика которых достаточно сложна. Многие биохимические критерии диагностики, предлагаемые для синдрома Марфана, неспецифич-ны для него, а определение оксипролина настолько трудоемко, что не может быть использова-но в исследованиях. Около 40 % больных имеют стертую форму заболевания, при которой отсутствуют выраженные изменения в какой-либо системе органов.
Большая часть работ в литературе на эту тему посвящена детям с синдромом Марфана, так как ранняя диагностика необходима для разграничения с фенотипически схожим наследст-венным заболеванием обмена метионина — гомоцистинурией, а также для своевременного меди-ко-генетического консультирования и прогнозирования будущего потомства.
Поздняя диагностика синдрома Марфана (у взрослых лиц) может свидетельствовать лишь о неудовлетворительном функционировании медико-генетической службы. Классический синдром Марфана не представляет больших затруднений для диагностики.
Целью работы было обобщение данных литературы и результатов собственных наблюдений болезни Марфана, выделение стертых форм ее, определение критериев диагностики, приведение рабочей классификации заболевания и решение вопросов динамического наблюдения.
При наличии классической триады симптомов и семейного характера заболевания диаг-ноз в большинстве случаев не вызывает сомнений, но, учитывая разноплановость этих измене-ний, одному специалисту не всегда удается заподозрить данное заболевание.
Клинические признаки синдрома Марфана


Примечание. + наличие, — отсутствие данного признака.
Под нашим наблюдением находились б мужчин в возрасте от 18 до 20 лет. Поводом для госпитализации послужили усиление кардиалгий, одышки, появление быстрой утомляемости при физических нагрузках. 3 человека поступили в отделение торакальной хирургии с диагнозом: воронкообразная грудная клетка с нарушением функции дыхания; 2 — в кардиологическое отде-ление с нейроциркуляторной дистонией по кардиальному типу; 1 больной острым бронхитом — в пульмонологическое отделение. Исходя из медицинских документов, все они считались здоро-выми. Таким образом, на догоспитальном этапе заболевание соединительной ткани — синдром Марфана — распознано не было. Диагноз верифицирован с помощью клинических и инструмен-тальных (рентгенография опорно-двигательного аппарата, ультразвуковое исследование сердца) методов с привлечением специалистов (отоларинголога, окулиста, невропатолога, нейро-хирурга, травматолога).
Данные о клинических и инструментальных признаках каждого больного с синдромом Марфана представлены в таблице. Учитывая эти данные, а также результаты исследований дру-гих авторов, критериями отбора лиц с возможным синдромом Марфана следует считать:
1. Семейный анамнез (наличие родственников с синдромом Марфана).
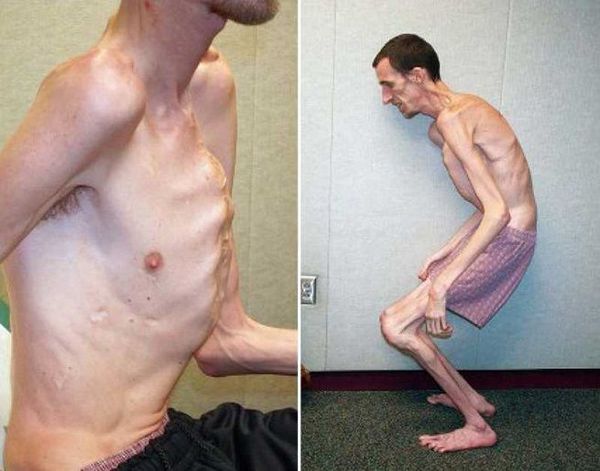
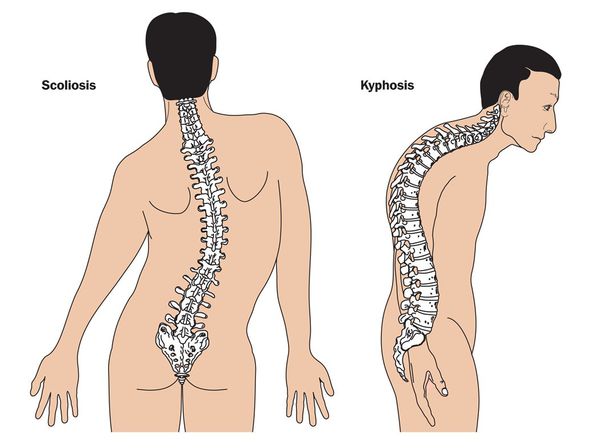
2. Изменение опорно-двигательного аппарата (высокий рост, чаще выше 190 см), несо-ответствие между ростом и массой тела в сторону ее уменьшения, длинные тонкие конечности, особенно с выраженным удлинением дистальных отделов, узкие кисти с длинными пальцами (арахнодактилия), плоскостопие, долихоцефалия, грыжи, идиопатические сколиозы, остеохонд-роз позвоночника, часто с грыжами Шморля, деформации грудной клетки, совокупность пере-численных изменений.
3. Нарушения со стороны глаз (подвывих хрусталика, высокая степень миопии, миопи-ческий астигматизм).
4. Изменения со стороны сердечно-сосудистой системы. Клинические: пролабирование митрального и других клапанов, недостаточность аортального клапана, быстро прогрессирую-щие кардиалгии неуточненной этиологии, особенно у лиц молодого возраста, аневризма аорты, особенно у молодых людей. Выявляемые специальными методами исследования: эхокардиография: пролабирование митрального клапана, других клапанов, нескольких клапанов; расширение кольца аорты и диаметра аорты, ее усиленная пульсация, признаки регургитации. Патолого-анатомически: аневризмы разных отделов аорты, разрыв аорты без анамнеза аортальной гипер-тензии у лиц без признаков атеросклероза аорты. Гистологически: эрдхеймовский некроз.
5. Изменения со стороны органов дыхания: спонтанный пневмоторакс, поликистоз лег-ких, ранние идиопатические формы эмфиземы, буллезная эмфизема. Рентгенологически: широкие межреберные промежутки, деформированные ребра, грудина, сколиоз.
6. Нарушения со стороны желудочно-кишечного тракта: висцероптоз, большое количест-во мукоидных веществ в желудочном содержимом.
7. Изменения ЛОР-органов и полости рта: готическое небо у лиц с узкими челюстями, неправильный прикус, уменьшение толщины костей черепа.
8. Рентгенологические находки: тонкие длинные трубчатые кости с поперечной исчер-чен-ностью и остеопорозом метафизов, изменение размеров передней черепной ямки, удлинен-ный канал спинного мозга, длинные тонкие кости кистей, стоп.
В отношении критериев диагноза существуют определенные разногласия между исследо-вателями. Ранее достоверными считались лишь случаи с выраженными проявлениями синдрома — сочетанием изменений в трех системах, которые у ряда пациентов могут наблюдаться уже в детском возрасте.
Однако большинство авторов, учитывая генетическую природу заболевания и особенно-сти характеристики доминантного гена, признают наличие неполных форм заболевания ив спорадических случаях. Биохимические критерии (изменения содержания глюкозаминогли-канов, оксипролина) важны, но, к сожалению, трудоемкость соответствующих анализов и не-возможность их осуществления в большинстве лечебных учреждений снижают их практическую ценность.
Доступными тестами для определения арахнодактилии являются:
— тест большого пальца, предложенный в 1945 г. (Parker, Haze, 1945): разогнутый первый палец приводится к кисти. Если тест положительный, палец выступает за мягкие ткани кисти. В сомнительных случаях необходима рентгенография кисти с приведенным большим паль-цем — фаланга большого пальца при положительном тесте выступает за скелет метакарпальных костей;
— тест запястья: при обхватывании пальцами растянутой кисти одной руки"запястья другой при арахнодактилии первый и пятый пальцы легко соединяются друг с другом.
Другие тесты, которые можно использовать для диагностики синдрома Марфана:
— соотношение кисть — рост более 11 %;
— соотношение ступня — рост более 15 %;
— размах рук больше роста на 5 см;
— длина среднего пальца больше 10 см;
— расстояние от лобка до пола превышает '/г роста более чем на 6 см.
Рентгенологический тест: метакарпальный индекс больше 8,4.
Если размах рук превышает рост более чем на 5 см, длина ног от лобка до пола более '/г роста, метакарпальный индекс больше 8,4, а также положителен результат теста большого пальца, диагноз синдрома Марфана следует считать достоверным.
Достоверность диагноза увеличивается при обследовании семьи.
Часто среди молодых людей выявляют кифосколиозы, некоторую гипермобильность суста-вов, плоскостопие, повышенную растяжимость связок, высокое небо, голубые склеры в сочета-нии с миопией, пролапс митрального клапана, мягкие ушные раковины. Комбинация этих при-знаков различна, так же как и степень их выраженности. О. В. Лисиченко относит таких больных к марфаноподобному варианту.
Таким образом, следует выделять следующие клинические варианты болезни:
1. Болезнь Марфана (наличие классических трех признаков, семейный характер заболе-вания).
2. Синдром Марфана (наличие стертых форм с положительными вышеперечисленными диаг-ностическими тестами).
3. Марфаноподобный синдром. Классификация болезни (синдрома) Марфана
(О. В. Лисиченко, 1986).
I. Форма
Стертая: слабо выраженные изменения в одной, двух системах. Выраженная:
1. Слабо выраженные изменения в трех системах.
2. Выраженные изменения хотя бы в одной системе (ограниченная форма).
3. Выраженные изменения в двух, трех системах и более.
Степень выраженности: а) легкая; б) средняя; в) тяжелая.
II. Характер течения
Рецидивирующий (прогрессирующий). Стабильный
III. Генетическая характеристика
Семейная форма (тип наследования). Первичная мутация.
Примерные диагнозы:
— болезнь Марфана, выраженная форма с преимущественным поражением сердечно-сосудистой системы средней степени тяжести, прогрессирующее течение, семейный случай, аутосомно-доминантный тип наследования:
— синдром Марфана, выраженная форма с преимущественным поражением глаз (подвывих хрусталика, миопатический астигматизм), сердечнососудистой системы (пролабирование мит-рального клапана с регургитацией (-|—(-), Hi), опорно-двигательного аппарата (долихосте-номе-лия, остеохондроз грудного отдела позвоночника без нарушения функции), дыхательной системы (диффузная эмфизема с нарушением функции внешнего дыхания I степени), спорадиче-ская форма;
— марфаноподобный синдром с преимущественным поражением опорно-двигательного аппа-рата (долихостеномелия, остехондроз грудного отдела позвоночника без нарушения функции), дыхательной системы (диффузная эмфизема легких с нарушением функции внешнего дыхания I степени), спорадическая форма;
— марфаноподобный синдром с преимущественным поражением опорно-двигательного аппа-рата (кифосколиоз, плоскостопие II степени), глаз (миопия высокой степени), сердечно-сосудистой системы (пролабирование митрального клапана без регургитации).
Вероятность правильного диагноза повышается при наличии совокупности признаков. В связи с этим необходимо оценивать значимость каждого признака с выяснением семейного анамнеза.
Диагноз выраженной формы при легкой степени изменений 3—4 систем и тем более диаг-ноз стертых форм правомочны в настоящее время, по мнению исследователей, только для род-ственников больных.
Со стороны сердечно-сосудистой системы к легким изменениям можно отнести начальную стадию пролабирования митрального клапана, к выраженным — пролабирование митрального и трех-куспидального клапанов, расширение аортального кольца, к тяжелым — аневризму аорты.
К легкой степени изменений опорно-двигательного аппарата относятся нарушения про-порций, воронкообразная деформация грудной клетки и сколиоз I степени; к средней степени — воронкообразная деформация, остеохондроз позвоночника с грыжами Шморля, сколиоз II сте-пени, плоскостопие, грыжи; к тяжелой — кифосколиоз III—IV степени, воронкообразная дефор-мация III—IV степени.
Со стороны глаз к изменениям легкой степени можно отнести гетерохромию радужки, малую степень миопии; к выраженным — подвывих хрусталика, миопический астигматизм, миопию высокой степени; к тяжелым — наличие осложнений в виде вывиха хрусталика, отслойки сет-чатки.
На основании собственных наблюдений и обобщенных данных литературы нами выделены и систематизированы (см. таблицу) наиболее частые признаки стертых форм синдрома Марфана, совокупность которых поможет упростить клинический, вариант дифференциальной диагностики.
При подозрении на синдром Марфана целесообразно использовать перечисленные в таб-лице изменения в комплексе. Критерии диагноза для родственников могут быть менее жестки-ми, когда родословная становится дополнительным, а иногда и решающим тестом для уточнения диагноза.
Лица с подозрением на синдром Марфана подлежат диспансерному динамическому наблю-дению с осмотром терапевтом через каждые 6 мес и другими специалистами (по показаниям), а также последующему (после постановки диагноза) медико-генетическому консультированию для лиц репродуктивного возраста.
Правильная диагностика и интерпретация клинических данных позволяют в ряде случаев уточнить диагноз синдрома Марфана, оценить функциональное состояние органов и систем, определить риск возможных осложнений, а поэтому играют существенную роль в распознавании заболевания в молодом возрасте и разработке профилактических мероприятий.
ЛИТЕРАТУРА
1. Бочкова Д. #., Артамонова Н. П., Кузьмина П. И. и др. Пролапс митрального клапана как симптом наследственных заболеваний // Вопр. охр. мат.— 1979.— № 10.— С. 12—18.
2. Гарпузов В. В. Клинико-биохимическое изучение синдрома Марфана: Автореф. дис. . канд.— Л., 1973.
3. Гордое И. Б., Ляхер А. В. Поражение сердечно-сосудистой системы при синдроме Марфана // Клин. мед.— 1980,— № 8.— С. 98—100.
4. Гофман В. А., Коробейникова С. А., Могилевский Р. Э. О клинической симптоматике стертых форм синдрома Марфана // Там же.— 1979.— № 6.— С. 90—92.
5. Казначеева В. П., Лисиченко О. В. Клинико-генетиче-ское обследование больных с синдромом Марфана. // Научная конф. по клинической генетике: Материалы.— М., 1971.—С. 9—11.
6. Лисиченко О. В. Синдром Марфана.— Новосибирск: Наука, 1986.
7. Наследственные системные заболевания скелета / Волков М. В., Меерсон Е. М., Негволдова О. Л. и др.— М.: Медицина, 1982.
8. Семячкина А. Н. Принципы диагностики синдрома Марфана: Автореф. дис. . канд.— М., 1975.
Обратите внимание, что эффективность средств народной медицины научно не доказана. Информация, размещенная на этой и иных страницах данного сайта, предназначена исключительно для ознакомления и обсуждения с врачом.
Обязательно проконсультируйтесь с врачом перед лечением.
Что такое синдром Марфана? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровиковой О. И., гинеколога-эндокринолога со стажем в 11 лет.

Определение болезни. Причины заболевания
Синдром Марфана (Marfan; СМ) — генетически обусловленное заболевание, при котором происходит системное поражение соединительной ткани. [1]
Этиологией заболевания является мутация в гене FBN1 (фибриллина 1), расположенном в коротком плече пятнадцатой хромосомы в локусе 21.1. [2]
Наследование заболевания происходит по аутосомно-доминантному типу, характеризуется высокой пенетрантностью (частотой появления гена) и различной экспрессивностью. [5]
Соотношение представителей мужского пола и женского одинаковое.
Симптомы синдрома Марфана
Наблюдается постоянно прогрессирующее развитие заболевания. У новорожденных детей выявляются удлинённые тонкие пальцы на верхних и нижних конечностях и удлинённые тонкие конечности (долихостеномелия). [1] У таких пациентов, помимо долихостеномелии, отмечается:
- повышенное физическое развитие;
- недостаток веса;
- удлинённый череп;
- вытянутое лицо;
- арахнодактилия (аномально удлинённые узкие пальцы);
- слабость и недоразвитие мышечной системы и жировой клетчатки;
- неловкие движения. [3]
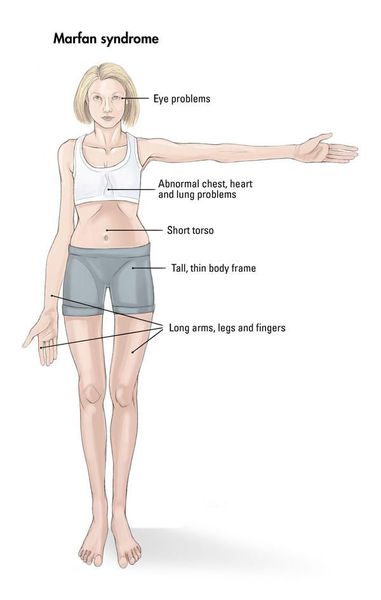
Кожа имеет повышенную растяжимость, разболтанные суставы. У большинства больных наблюдается высокое аркообразное нёбо, изменения формы грудной клетки (воронкообразная, килевидная) и искривления позвоночника (сколиоз в 60%, кифоз (изгиб позвоночника с образованием горба), ювенильный остеохондроз), уплощение свода стопы, аускультативные признаки порока сердца (шумы). [4] Длина третьего пальца руки — 10 см и больше (скрининговый тест у детей 7-18 лет): возрастает соотношение размаха верхних конечностей к длине тела.
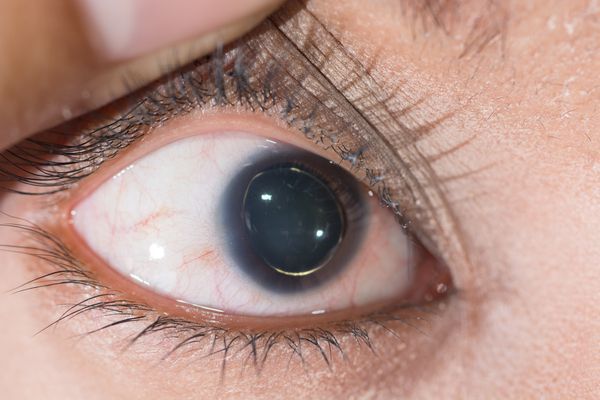
Офтальмологические симптомы (близорукость, подвывих хрусталика в 75% случаев, его округлость или гипоплазия, отслойка сетчатки) и астенические признаки (усталость, вялость) обращают на себя внимание со второго года жизни, изменения формы грудной клетки появляются в возрасте старше четырёх лет, патология сердца и сосудов выявляется в дошкольном возрасте. [1]
Почти у всех больных выявляются пороки сердца и аорты. Часты бедренные и паховые грыжи, поражение клапанов в венах, их варикозное расширение, геморрагический синдром, рецидивирующие вывихи, поражение лёгочной системы (самопроизвольный пневмоторакс, эмфизематозное расширение лёгких), опущение почек. [2]
В четверти случаев зарегистрировано снижение интеллекта, у половины пациентов выявляются нарушения эмоционально-волевой сферы. Часто появляются депрессивные состояния, нейроциркуляторная дистония. [3]
По данным многих исследований, абсолютное большинство больных с синдромом Марфана отмечают ухудшение эмоционального фона, утрату чувства радости и увлечённости профессиональной деятельностью, частую смену настроения, повышенную возбудимость, чувство тревоги. Результатом этого является снижение социальной активности, ухудшение качества жизни и значительное уменьшение социальной адаптации. [3]
У таких пациентов часто наблюдается трахеобронхиальная дискинезия (нарушение дыхательной системы) за счёт слабости соединительнотканного каркаса бронхов. Это проявляется рецидивирующими воспалительными заболеваниями бронхолегочной системы, обструктивными нарушениями, бронхиальной астмой, эмфиземой лёгких (повышенное содержание воздуха в лёгочной ткани). [4] Встречаются осложнения, которые проявляются скоплением воздуха в грудной клетке, сопровождающиеся сдавлением лёгких и средостения (срединной области грудной клетки), подкожной эмфиземой. Наблюдается неадекватный ответ на бронхолитики. Обструктивные явления (непроходимость) затрагивают преимущественно верхние отделы респираторного тракта. [3]
Описаны характерные изменения на электрокардиограмме, включающие синдром раннего возбуждения желудочков, преждевременные желудочковые комплексы, нестабильность конечной части желудочкового комплекса в задненижних отведениях. [3]
Патология ритма чаще всего проявляются блокадой правой ножки пучка Гиса или смешанной экстрасистолией. [6]
У больных синдромом Марфана с патологией ритма сердечной деятельности и проводимости синдром вегетативной дисфункции чаще протекает по ваготоническому типу, в виде пресинкопальных, обморочных и астеновегетативных состояний, болезненных ощущений в области сердца, цефалгии напряжения (головной боли) и зачастую сочетается с психопатологическими расстройствами. [4]
Органы пищеварения также задействованы в патологическом процессе, что проявляется дискинезией (нарушением моторики) билиарного тракта со снижением моторики гладкомышечной мускулатуры, недостаточностью кардии, грыжевыми выпячиваниями пищеводного отверстия диафрагмы, аномалиями желчевыводящих протоков, долихосигмой (увеличением сигмовидной кишки), хроническим гастродуоденитом (воспалением слизистой желудка и двенадцатиперстной кишки), дисбиозом (нарушением нормальной микрофлоры) кишечника, изменениями поджелудочной железы. [3]
У пациентов с синдромом Марфана чаще, чем у здоровых людей, встречаются приобретённые аномалии почек: повышенная подвижность почек, нефроптоз (опущение почки), пиелоэктазии (аномальное расширение лоханок), повышена частота удвоения почек.
Патогенез синдрома Марфана
Более половины веса человека представлено соединительной тканью, из неё состоит наша главная опора — скелет, внешние покровы — кожа. Сосуды, кровь и лимфа тоже состоят из соединительной ткани.
К клеткам соединительной ткани относятся фибробласты и их разновидности (остеобласты, хондроциты, одонтобласты, кератобласты), макрофаги (гистиоциты) и тучные клетки (лаброциты). [7]
Мезенхима — проводник конституциональных, генетических и эпигенетических составляющих жизни человека. Патология соединительной ткани детерминирует определенное патологическое действие на весь организм в целом, на его физиологию и его конституциональные особенности. [3]
При болезни Марфана происходит замена нуклеотидов в гене, содержащем информацию о структуре пептида фибриллина-1. Этот белок относится к гликопротеидам, принимает участие в микрофибриллярном комплексе, он обеспечивает основу эластических фибрилл соединительной ткани.
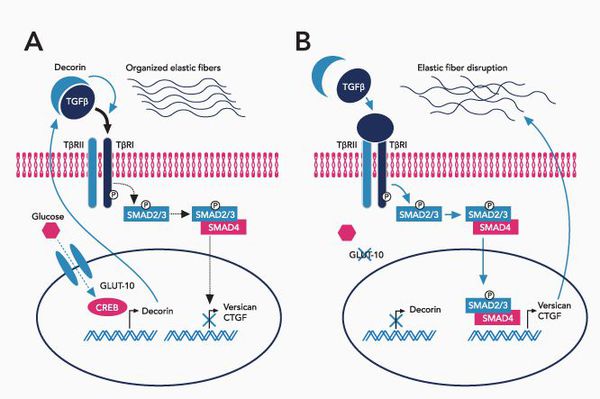
Межклеточный матрикс позволяет соединительной ткани поддерживать постоянную структуру, в нём находится огромное количество факторов роста, которые обеспечивают постоянное обновление клеток.
В крупных сосудах, связочном аппарате содержится большое количество эластиновых фибрилл, поражение которых и даёт основные клинические проявления синдрома Марфана.
При синдроме Марфана значительно поражается трансформирующий фактор роста бета (TGF-β), нарушается связывание его неактивной формы, что приводит к повышению биоактивности данного фактора, с чем связано появление многих проявлений болезни. [4]
Патология фибриллина приводит к патологии формирования волокон, что вызывает утерю прочности и эластичности кожи и других соединительнотканных структур.
Изменение структуры коллагеновых волокон приводит к нарушению первичного звена гемостаза у пациентов с синдромом Марфана. [6]
Имеются данные о дефектах мембранных и цитоплазматических механизмов проведения сигнала непосредственно в самом тромбоците, приводящих к нарушениям агрегации (объединения). Показано наличие самостоятельного мембранного дефекта тромбоцитов, протекающего с нарушением реакций высвобождения и транспорта внутриклеточного кальция. [6]
Отмечена роль гормонального дисбаланса в развитии и усугублении дефектов соединительнотканных структур. [3]
Тромботические проявления детерминированы нарушением реологии (вязкости) крови в патологически извитых сосудах брахиоцефальной зоны. [3]
Поражение желудочно-кишечного тракта детерминировано тем, что эта система богата коллагеном. Наблюдаются дискинезия билиарного тракта по гипомоторному типу, грыжи пищеводного отверстия диафрагмы, аномалии желчных путей, долихосигма, хронический гастродуоденит со стёртой клинической картиной, склонностью к торпидному течению. [3]
Классификация и стадии развития синдрома Марфана
- стёртая (поражено не более двух систем, изменения выражены незначительно);
- выраженная (незначительные изменения в трёх системах либо значительное поражение одной и более систем).
Выделяют различные типы по степени тяжести:
- лёгкая
- средняя
- тяжёлая
Частота тяжёлых форм — 1 к 25000-50000 (при общей частоте диагностированных случаев 1 к 10000-15000).
По характеру течения:
- прогрессирующая форма;
- стабильная форма.
Чаще всего первые признаки синдрома Марфана проявляются еще в детском периоде, с возрастом происходит прогрессирование симптомов, усиление клинических проявлений.
Осложнения синдрома Марфана
К самым частым осложнениям синдрома Марфана относятся:
- Снижение зрения, вплоть до слепоты, обусловленное слабостью цинновой связки (ресничного пояска) и подвывихом, вывихом хрусталика. [7]
- Сердечная недостаточность по застойному типу, обусловленная нарушением сократимости сердечной мышцы, недостаточностью митрального клапана. [6]
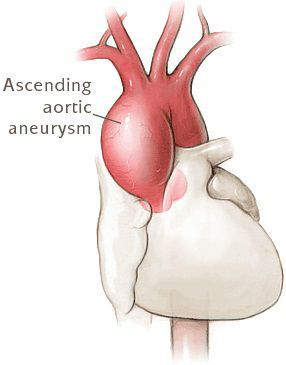
- Разрывы крупных сосудов, связанные с дилатацией (расширением), истончением стенки сосудов. Чаще всего происходит поражение аорты (в основном из-за изменения гемодинамики при беременности). [7]
- Расслаивающая аневризма аорты, приводящая к смерти больных.
Диагностика синдрома Марфана
Диагностика Синдрома Марфана основывается на клинических данных, выявлении изменений в гене FBN1. [5]
Часто при сборе генеалогического анамнеза выявляются родственные случаи со скрытым течением заболевания. [1]
Способы обнаружения арахнодактилии: [3]
- Симптом Steinberg (признак первого пальца). Первый палец виден из-под hypothenar при напряжённом кулаке.
- Симптом Walker-Murdoch (признак запястья). При обхватывании кистью в области лучезапястного сочленения контралатеральной верхней конечности первый палец заходит за пятый.
- Определение пястного индекса. Определяется при помощи рентгенографии. Средняя длина пясти, делённая на усреднённую ширину отрезка от второй до четвертой пястной кости. При нормальном соотношении этот показатель соответствует 5,4-7,9, в то время, как при синдроме Марфана — больше 8,4.
В 2010 году группа специалистов систематизировала международные Гентские критерии для верификации синдрома Марфана. Верификация зависит от данных генеалогического анамнеза. [3]
При отсутствии генеалогического анамнеза:
- увеличение диаметра аорты >, = 2 ϭ + эктопия хрусталика = СМ;
- увеличение диаметра аорты >, = 2 ϭ + выявленные изменения в гене FBN1 = CM;
- увеличение диаметра аорты >, = 2 ϭ + >, = 7 системных признаков = СМ;
- эктопия хрусталика + наличие изменений в гене FBN1 + дилатация аорты = СМ;
При наличии генеалогического анамнеза:
- Эктопия хрусталика + случай СМ в семье = СМ;
- >, = 7 системных проявлений + случай СМ в семье = СМ;
- увеличение диаметра аорты >, = 2 ϭ + случай СМ в семье = СМ.
В пятнадцати процентах появление ребёнка с синдромом Марфана спорадическое (случайное), у родителей могут быть слабые проявления. У родственников пациентов встречаются заболевания желудочно-кишечного тракта, поражения позвоночника, заболевания глаз. [3]
При малейшем подозрении на синдром Марфана необходима консультация офтальмолога. В анализе мочи таких пациентов отмечается повышение уровня оксипролина, гликозаминогликанов, но эти показатели низкоспецифичны, могут быть при различных дисплазиях соединительной ткани. Выделение оксипролина является показателем тяжести заболевания. Наблюдается нарушение свертываемости крови на тромбоцитарном уровне. [3]
Оценка системных признаков вовлечённости соединительной ткани
Существуют заболевания, причина которых скрыта в генетическом дефекте, а их проявления выражаются в сочетании определённых симптомов. Эти болезни ещё называют наследственными синдромами. Науке известно более 6000 генетических недугов, многие из которых встречаются крайне редко. Но некоторые заболевания известны врачу любой специальности, к таким болезням относится синдром Марфана.
Пациентов с этим диагнозом можно увидеть на приёме у разных специалистов, поскольку болезнь протекает с поражением многих органов и систем. Родителям необычных детей нужно знать, как проявляется этот недуг, и какие действия помогут улучшить качество жизни ребёнка.
О заболевании
Синдром Марфана – генетический недуг, который выражается в недоразвитии соединительной ткани. Заболевание относится к редким, по статистике наследственная болезнь обнаруживается у 1 из 10000 детей. Возникновение синдрома не зависит от расовой принадлежности и пола малыша, и с одинаковой частотой проявляется, как у мальчиков, так и у девочек различных национальностей.
Историческая справка
Первые упоминания о необычном недуге можно обнаружить в трудах американского офтальмолога Э. Вильямса, который в 1875 году описал признаки идентичного смещения хрусталиков глаз у родных брата и сестры. Кроме офтальмологических проблем эти дети имели повышенную подвижность суставов и высокий рост.
Известность болезнь приобрела позже, через 20 лет, когда французский педиатр Антуан Марфан представил свои наблюдения за 5-тилетей больной. Маленькая пациентка отличалась необычными аномалиями скелета и быстрым прогрессированием недуга. Синдром был назван в честь французского доктора, хотя впоследствии стало известно, что наблюдаемая им девочка страдала другой наследственной патологией – врождённой контрактурной арахнодактилией.
Почему проявляется генетический синдром?
Причиной развития недуга считается мутация в гене FBN1, который располагается в 15 хромосоме и отвечает за нормальное производство фибриллина 1. Этот белков соединительной ткани является одним из главных компонентов, придающих ей эластичность и способность к сокращению.
Первыми при генетическом синдроме поражаются структуры, содержащие наибольшее количество важного белка – стенки кровеносных сосудов, связочный аппарат, цинновая связка глаза. Изменённая соединительная ткань не способна выполнять своей функции, выдерживать физическую нагрузку в связи с потерей прочности и упругости, у ребёнка возникают симптомы заболевания.
Недуг относится к генетическим и передаётся от родителей по аутосомно-доминантному типу. Риск появления малыша с наследственным синдромом очень высокий, если у мамы или папы имеются признаки болезни. В 75% случаях заболеваний прослеживается появление недуга в каждом поколении семьи. У 25% больных определяется новая, спонтанная мутация, не находится чёткой связи с наследованием.
Классификация синдрома
Болезнь отличается многообразием проявлением и различной их выраженностью. Этот недуг может быть длительное время нераспознанным, а некоторые отличительные особенности ребёнка расцениваться, как вариант нормы. В то же время существую формы, при которых характерные признаки болезни видны уже в роддоме.
В зависимости от выраженности клинических проявлений различают 2 формы болезни:
- стёртую.
Признаки поражения органов незначительные и охватывают 1 – 2 системы организма;
- выраженную.
Данную форму определяют, если хотя бы одна из систем организма имеет серьёзные нарушения функций. Врач может поставить этот диагноз и в случае обнаружения умеренных поражений 2 – 3 систем организма.
Большое значение в определении прогноза заболевание имеет динамика нарушений (специалисты различают прогрессирующий и стабильный варианты синдрома).
Классические симптомы заболевания
Некоторые проявления наследственной болезни можно обнаружить даже у новорождённых малышей, такие крохи имеют большую длину тела, чем их сверстники, длинные пальцы, умеренные поражения костной системы и внутренних органов. Но характерная симптоматика формируется к 7 – 8 годам жизни ребёнка, со временем признаки болезни становятся более выраженными, возникают новые проявления. Все больные синдромом Марфана имеют схожие поражения внутренних органов, кроме того их внешний вид так же типичный.
При рассмотрении лица больного можно заметить вытянутый овал, небольшую нижнюю челюсть, близко посаженные глаза. Возможен неправильный рост зубов, нарушенный прикус, а нёбо крохи расположено выше, чем у других детей.
Поражение соединительной ткани значительно сказывается на строении и функционировании костной системы. Позвоночник ребёнка не способен выполнять свою опорную функцию, выдерживать возрастающую с ростом малыша нагрузку на него.
Возникают различные деформации позвоночного столба – сколиоз, кифоз, их сочетание. Из-за нестабильности связочного аппарата возникают подвывихи и вывихи шейного отдела позвоночника.
Грудная клетка ребёнка также деформируется, возникают её смещение наружу (килевидная грудь) или наоборот, западение грудины (воронкообразная, впалая грудь). Также у больных детей нередко развиваются плоскостопие, рекурвируются колени (избыточно разгибаются в суставах).
Кроме того, у ребят наблюдается повышенная гибкость, гипермобильность суставов, что связано с растяжимостью хрящевой ткани, связок и суставов. Такие дети отличаются большей пластичностью, чем их сверстники. Нередко родители радуются этой особенности малыша, особенно если деформации скелета выражены незначительно, и решают отдать кроху в спортивную секцию.
Одним из ведущих признаков, определяющих течение и исход заболевания, является поражение сердца и сосудов. Дефект соединительной ткани проявляется патологией строения стенок крупных сосудов, клапанов и перегородок сердца.
В тяжёлых случаях малыш рождается с врождённым пороком сердца, угрожающим жизни и требующим незамедлительной хирургической коррекции. Нередко у детей выявляются признаки пролапса клапанов, чаще митрального и аортального. Из-за недостаточной эластичности соединительной ткани клапаны (своеобразные створки, которые препятствуют обратному току крови) не могут выполнить свою функцию. Возникают нарушения кровообращения, которые проявляются обмороками, головокружением, одышкой, повышенной утомляемостью.
Самым опасным состоянием при синдроме Марфана считается расширение, расслаивание стенки и разрыв аорты. Её поражение носит врождённый характер и имеет тенденцию к прогрессированию, принося всё новые опасные патологические симптомы. Поэтому наблюдение за состоянием сердечно-сосудистой системы и своевременное предотвращение и лечение осложнений – главная задача в терапии синдрома Марфана.
Другие патологии сердца при синдроме Марфана проявляются нарушением ритма и проводимости, развитием бактериальных осложнений – инфекционного эндокардита.
Около половины больных наследственным синдромом людей имеют эктопию хрусталика, которая развивается обычно до 4-хлетнего возраста и со временем прогрессирует. В норме хрусталик удерживается в правильном положении с помощью цинновых связок. У детей с патологией соединительной ткани возникает слабость связочного аппарата глаза, что приводит к развитию заболеваний.
Кроме того со стороны органов зрения возникают такие патологии, как близорукость, повышение внутриглазного давления – глаукома, отслойка сетчатки, колобома радужки и другие заболевания.
Поскольку соединительная ткань является составной частью любого органа, у ребёнка могут возникнуть разнообразные патологии. Со стороны нервной системы иногда возникает такая патология, как менингоцеле. Из-за врождённого дефекта позвоночника происходит выпячивание спинного мозга и его оболочек, обычно это наблюдается в пояснично-крестцовой области.
Дети с синдромом Марфана имеют слабо развитую мышечную ткань. Нередко у них возникают грыжевые выпячивания, которые рецидивируют, возникают снова даже после оперативного лечения. Часто наблюдается изменение положения внутренних органов – опущение почек, матки, мочевого пузыря.
Даже небольшие травмы и падения опасны для больных генетическим синдромом. Вывихи, разрывы связок, которые долго не заживают, часто приносят беспокойства малышу. Со стороны дыхательной системы отмечаются врождённые пороки развития лёгких, кистозные изменения ткани, спонтанные пневмотораксы (надрыв плевры и скопление воздуха в плевральной полости).
Диагностика синдрома Марфана
Для правильной постановки диагноза малышу придётся пройти комплексное обследование, получить консультацию у многих специалистов.
Поскольку патология имеет наследственную природу, в большинстве случаев удаётся проследить заболевание в семье. Нужно помнить, что недуг может протекать в различных формах и многие люди, с лёгким течение синдрома, не догадываются о своём заболевании на протяжении всей жизни.
- большие критерии.
К ним относятся:
- увеличение роста в большей степени за счёт верхней части тела;
- грубая деформация грудной клетки и позвоночника;
- продольное плоскостопие;
- невозможность полностью разогнуть конечность в коленных и локтевых суставах (контрактуры);
- эктопия хрусталика;
- расширение и расслоение восходящей части аорты и другие признаки.
- малые критерии.
Эти признаки в меньшей степени указывают на наследственный дефект, но их наличие и сочетание с большими критериями подтверждает диагноз синдром Марфана.
К ним относятся:
- высокая подвижность суставов;
- аномалии зубов, нёба;
- гипоплазия радужной оболочки глаза, цилиарной мышцы, увеличение длины глазного яблока;
- пролапс митрального клапана;
- патологии бронхо-лёгочной системы, спонтанный пневмоторакс и другие нарушения.
Для уточнения внешних изменений у ребёнка используются различные способы: измерение роста, длины кисти, соотношение размеров верхней части туловища к нижней и другие. Особо показательны в диагностике синдрома следующие исследования:
- тест запястья.
- тест большого пальца.
Исследователь просит малыша попытаться дотянутся большим пальцем до предплечья этой же руки. Тест считается положительным, если ногтевая фаланга пальца ребёнок с лёгкостью достаёт до лучевой кости предплечья.
Обычные клинические и биохимические анализы крови и мочи не показательны при синдроме Марфана, изменений в них может не быть. Помочь в установлении диагноза поможет обнаружение продуктов метаболизма соединительной ткани в моче.
Специфические проявления заболевания можно обнаружить во многих органах, поэтому заподозрив у ребёнка наследственный синдром, проводятся различные исследования. Патологию костно-суставной системы обнаруживают с помощью рентгенографии и компьютерной томографии.
Болезни сердца и сосудов диагностируются благодаря ЭКГ и ЭхоКС, МРТ. С помощью УЗИ обнаруживаются патологии внутренних органов, смещение их положения в брюшной полости. В исследовании органа зрения помогут такие методы, как офтальмоскопия, биомикроскопия.
Ребёнок с наследственным синдромом состоит на учёте у многих врачей: генетика, травматолога, хирурга, офтальмолога, кардиолога и других специалистов.
Лечение синдрома Марфана
Специфической терапии, направленной на устранение причины заболевания не существует. В настоящее время не разработаны методы влияния на наследственный аппарат клетки. Поэтому основная цель лечения синдрома Марфана – предотвращение прогрессирования заболевания, борьба с симптомами болезни:
- патология сердца и сосудов.
Самым опасным проявлением недуга считается аневризма, расширение участка аорты. Коварство болезни заключается в непрерывном, длительном прогрессировании симптомов. Случается, что опасный симптом формируется к 18 годам, поэтому важно уделять достаточно внимания ежегодному обследованию и лечению сердечно-сосудистой системы.
Грубые пороки развития сердца и сосудов, тяжёлые осложнения хронических болезней лечатся оперативно. Из лекарственных препаратов назначаются ингибиторы АПФ, блокаторы кальциевых каналов. Применение b-адреноблокаторов (пропанолола, атенолола) показано при расширении корня аорты, пролапсе клапанов, аритмиях.
- болезни опорно-двигательного аппарата.
Существую исследовании, указывающие на дефицит некоторых макроэлементов (кальций, цинк, кобальт, магний) и белков, необходимых для строительства соединительной ткани при синдроме Марфана. Поэтому для предотвращения прогрессирования патологии назначаются витаминно-минеральные комплексы, гиалуроновая кислота, викасол, колекальциферол. Оперативное лечение показано при грубых патологиях развития скелета;
- заболевания органов зрения.
Исправление патологии зрения проводится с помощью подбора специальных очков, контактных линз, оперативного лечения катаракты и глаукомы, смещения хрусталика.
- нарушенный обмен веществ.
Для улучшения метаболизма рекомендовано использовать в комплексном лечении аскорбиновую и янтарную кислоты, карнитин, препараты магния, токоферола ацетата. С целью нормализации обмена хрящевой ткани используются глюкозаминсульфат, хондроитинсульфат.
Прогноз и профилактика
Течение болезни во многом зависит от выраженности клинических проявлений и качества проведённого лечения. Особую опасность для жизни ребёнка представляют пороки развития сердца и сосудов, их патологические изменения, поэтому терапии этой группы заболеваний отводится особое место.
Родителям девочек нужно знать об опасности будущей беременности для здоровья больной наследственным синдромом. Именно во время вынашивания ребёнка увеличивается риск развития и расслаивания аневризмы аорты. Это связано с повышенной нагрузкой на систему кровообращения и гормональными изменениями.
В целом, при правильном лечении и вовремя оказанной помощи пациенты с генетическим синдромом доживают до глубокой старости. Болезнь привносит ограничения в выборе будущей профессии. Таким детям лучше искать занятие не связанное с повышенной физической нагрузкой, поднятием тяжестей.
Профилактика недуга заключается в своевременном диагностировании синдрома в семье и медико-генетическом консультировании будущих родителей.
Выводы
Синдром Марфана – сложное заболевание, которое характеризуется многообразием проявлений и различной их выраженностью. Дети с генетическим дефектом отличаются от своих сверстников и внешне, и проблемами со здоровьем. Родителям необычных детей важно понимать особенности своего малыша, обеспечить ему регулярное обследование и своевременное лечение патологий внутренних органов. Таким образом можно значительно улучшить качество жизни крохи и предупредить развитие опасных осложнений.
Читайте также:
