
Синдром коротких ребер что это
Рубрика МКБ-10: Q77.2
Содержание
- 1 Определение и общие сведения
- 2 Этиология и патогенез
- 3 Клинические проявления
- 4 Синдром короткого ребра: Диагностика
- 5 Дифференциальный диагноз
- 6 Синдром короткого ребра: Лечение
- 7 Профилактика
- 8 Прочее
- 9 Источники (ссылки)
- 10 Дополнительная литература (рекомендуемая)
- 11 Действующие вещества
Синдром короткого ребра c полидактилией
Синдром короткого ребра c полидактилией представляют собой группу костных пороков развития, характеризующихся узкой грудной клеткой и полидактилией (обычно преаксиальной).
Распространенность неизвестна. Все эти клинически различающиеся синдромы наследуются как аутосомно-рецессивные признаки.
Каузативные гены были идентифицированы для синдрома Эллиса- ван Кревелда - гены EVC и IFT80 ( 3q25.33).
Группа неоднородна и включает в себя относительно доброкачественные синдромы Жене и Эллиса-ван Кревелда, а также летальные хондродисплазии: тип 1 (Салдино-Нунана), тип 2 (Маевского), тип 3 (Верма-Наумофф) и тип 4 (Бимера-Лангера). Эта классификация основывается на рентгенологических данных.
Пороки развития внутренних органов отмечаются часто и отличаются между синдромами. Некоторые случаи заболевания не соответствуют какой-либо категории.
Асфиксическая дистрофия грудной клетки
Синонимы: синдром Жена
Определение и общие сведения
Асфиксическая дистрофия грудной клетки является формой дисплазии короткого ребра. Характеризуется узкой грудной клеткой, короткими конечностями и различными рентгенологическими скелетными аномалиями, включая "трезубец" вертлужной впадины и изменениями метафизов.
Точная ежегодная заболеваемость при рождении неизвестна, по оценкам 1-5 / 500000.
Синдром передается как аутосомно-рецессивный признак. Риск рецидива составляет 25% для каждой беременности после рождения больного ребенка.
Этиология и патогенез
Молекулярная основа синдрома была частично выяснена и указывает на причастность генов IFT80 (3q25.33), DYNC2H1 (11q22.3), WDR19 (4p14) и TTC21B (2q24.3), каждый из которых кодирует интрафлагеллярный транспортный белок. Таким образом синдром Жена принадлежит к группе цилиопатий. Мутации в других, еще не идентифицированных генах, также могут быть причиной патологии.
Клинические проявления
Синдром Жена проявляется в дородовый период или при рождении. В редких случаях может также присутствовать постаксиальная полидактилия. Узкая грудная клетка может способствовать развитию неонатальной дыхательной недостаточности, а также может быть причиной постоянных респираторных заболеваний. Некоторые случаи асфиксической дистрофии грудной клетки протекают тяжело, другие имеют доброкачественное течение. Рост пациентов варьирует, но может быть близким к норме. Сообщалось о редких случаях развития печеночной и почечной недостаточности, которые проявлялись фиброзом печени или нефронофтизом и отмечались в любом возрасте. Также наблюдалась ретинальная пигментная дегенерация. Интеллектуальное развитие нормальное.
Диагностика
Диагноз основывается на рентгенологических признаках: короткие ребра, таз имеет аномальную морфологию - с горизонтальной вертлужной крышкой и трезубцем, образованным медианным выступом и двумя боковыми отростками. Верхние конечности нормальные или укорочены, возможны конусообразнын эпифизы фаланг пальцев.
Тщательное дородовое ультразвуковое исследование может выявить заболевание.
Дифференциальный диагноз
Дифференциальный диагноз должен включать тораколарингопельвическую дисплазию (синдром Барнса), синдром Эллиса-ван Кревельда, синдром Сенсенбреннера и однородительская дисомия 14 хромосомы.
Лечение состоит в контроле над респираторными инфекциями. Следует регулярно контролировать функции почек и печени, а также состояние сетчатки глаз.
Прогноз сильно варьирует в зависимости от сопутствующих заболеваний внутренних органов. Риск тяжелых респираторных осложнений снижается после возраста 2-х лет.
Это группа наследственных заболеваний опорно-двигательного аппарата, которые часто проявляются карликовостью и нарушениями пропорций тела.
У некоторых больных рост и телосложение нормальные, но имеются характерные изменения глаз или расщелина неба, часто встречающиеся и у более тяжелых больных.

Нередко развиваются дегенеративные изменения суставов, поэтому легкие формы хондродисплазий у взрослых трудно отличить от генерализованного деформирующего остеоартроза.
Классификация
Выделено более 150 типов хондродисплазий, составляющих 8 основных групп.
Группа ахондроплазии
- Ахондроплазия
- Гипохондроплазия
- Танатофорная дисплазия
Спондилоэпифизарные дисплазии
- Ахондрогенез, тип IA (тип Хьюстона—Харриса)
- Ахондрогенез, тип IB (тип Фраккаро)
- Ахондрогенез, тип II (тип Пантера—Салдино)
- Гипохондрогенез
- Врожденная спондилоэпифизарная дисплазия Спондилоэпиметафизарная дисплазия, тип Страдвика (синдром Страдвика)
- Синдром Стиклера
- Дисплазия Книста
Точечные хондродисплазии
- Ризомелическая точечная хондродисплазия
- Точечная хондродисплазия Конради—Хюнермана (аутосомно-доминантная)
- Х-сцепленная доминантная точечная хондродисплазия
- Х-сцепленная рецессивная точечная хондродисплазия
Синдромы коротких ребер
- Хондроэктодермальная дисплазия (синдром Эллиса—ван Кревельда)
- Асфиктическая дисплазия грудной клетки
- Синдром коротких ребер, тип I (синдром коротких ребер — полидактилии, тип Салдино—Нунан)
- Синдром коротких ребер, тип II (синдром коротких ребер — полидактилии, тип Маевского)
Метатропные дисплазии
- Метатропная дисплазия
- Легкие формы метатропной дисплазии
Метафизарные хондродисплазии
- Метафизарная хондродисплазия, тип Янсена
- Метафизарная хондродисплазия, тип Шмида
- Метафизарная хондродисплазия, тип Мак-Кьюосика
Брахиолмия
- Брахиолмия I типа
- Брахиолмия II типа
- Брахиолмия III типа
Периферические дизостозы
- Периферический дизостоз
- Акродизостоз
- Трихо-рино-фалангеальный синдром, тип I
- Трихо-рино-фалангеальный синдром, тип II (синдром Лангера— Гидиона)
Выделяют хондродисплазии, приводящие к смерти (танатофорные), вызывающие искривление костей (диастрофические), поражающие метафизы (метафизарные), поражающие эпифизы (эпифизарные) и другие.
Некоторые хондродисплазии названы по имени первого или наиболее подробно описанного больного.
Тяжелые формы хондродисплазий сопровождаются выраженной деформацией большинства хрящевых структур и поражением глаз.
Легкие формы классифицировать труднее. Основные симптомы хондродисплазий — катаракта, дегенерация стекловидного тела, отслойка сетчатки, высокий лоб, гипоплазия лицевых костей, расщелина неба, короткие тонкие конечности и выраженная деформация эпифизов, метафизов и суставных поверхностей.
Распространенность
Хотя точная распространенность большинства хондродисплазий неизвестна, возможно, что это одни из самых частых наследственных болезней соединительной ткани: распространенность синдрома Стиклера типа I (артроофтальмопатии), например, достигает 1:10 000.
Молекулярные нарушения
Первые мутации при хондродисплазиях были выявлены в гене COL2A1, кодирующем коллаген II типа — основного белка хрящевой ткани. Всего в этом гене выявлено более 40 мутаций, вызывающих различные хондродисплазии.
Мутации гена COL2A1 обнаружены примерно у 20% больных с тяжелыми и среднетяжелыми хондродисплазиями и у 2% больных с ранним генерализованным деформирующим остеоартрозом. Сходная клиническая картина может быть вызвана также мутациями других генов: трех других типов коллагена, входящих в состав хряща; факторов роста; рецепторов факторов роста; факторов транскрипции.
Число описанных мутаций не отражает реальной генетической гетерогенности той или иной болезни: оно зависит от сложности структуры гена, технических трудностей, доступности больших семей для генодиагностики и интенсивности исследовательской работы. Очевидно, будут выявлены новые мутации.
Мутации гена COL2A1 приводят к дефектам проколлагена II типа. Большинство из 40 известных таких мутаций вызывают тяжелые хондродисплазии, такие, как ахондрогенез типа II (тип Лангера—Салдино) и дисплазия Книста. Однако мутации этого гена обнаружены и в некоторых семьях, члены которых в детстве были здоровы, но в зрелом возрасте у них появились симптомы деформирующего остеоартроза (боль, скованность и дегенеративные изменения суставов).
Наследственные дефекты проколлагена II типа сходны с дефектами проколлагенов I и III типов, и при них так же трудно установить соответствие генотипа и фенотипа. Тем не менее известно, что мутации, вызывающие образование терминирующего кодона и синтез укороченного белка, приводят к синдрому Стиклера типа I (артроофтальмопатии).
При деформирующем остеоартрозе с легкой формой хондродисплазии обнаружены мутации, приводящие к замене аргинина на цистеин в положении Y повторяющейся последовательности -Гли-X-Y-.
При метафизарной хондродисплазии (тип Шмида), которая проявляется низкорослостью, Х-образным искривлением ног, искривлением метафизов и утиной походкой, обнаружены дефекты коллагена X типа, короткие молекулы которого синтезируются в основном хондроцитами II типа и образуют сеть в межклеточном веществе хряща. Некоторые формы синдрома Стиклера (тип III, то есть без поражений глаз) вызваны дефектами α2(Х1)-цепи коллагена XI типа, малораспространенного коллагена, входящего в состав хряща и других тканей.
У большинства больных ахондроплазией (это самая частая причина непропорциональной низкорослости, сопровождающейся макроцефалией и дисплазией метафизов длинных трубчатых костей) обнаружены мутации гена FGFR3, кодирующего рецептор фактора роста фибробластов.
Замена лишь одного нуклеотида, вызывающая замену глицина на аргинин в положении 380 молекулы рецептора, имеется почти у 90% больных. Обычно это новые мутации, так что эта точечная мутация — одна из наиболее часто возникающих мутаций в геноме человека. Она приводит к конститутивной (не зависящей от лиганда) активации рецептора и нарушению развития хряща.
При более тяжелых болезнях, таких, как гипохондроплазия и танатофорная карликовость, а также в некоторых семьях с краниосиностозом мутации затрагивают другие участки гена FGFR3. Однако у большинства больных с краниосиностозом обнаруживают мутации родственного гена FGFR2.
При множественной эпифизарной дисплазии и псевдоахондроплазии, родственных болезнях, проявляющихся укорочением конечностей и деформирующим остеоартрозом, обнаружены мутации гена СОМР, кодирующего олигомерный белок хрящевого матрикса. Однако в одной семье с эпифизарной хондродисплазией обнаружена мутация гена COL9A1, кодирующего а2(1Х)-цепь коллагена IX типа.
Диагноз при тяжелых формах хондродисплазий ставят на основании характерной внешности, рентгенологических данных, гистологических изменений и течения болезни. Иногда, хотя и реже, чем при несовершенном остеогенезе, оказывается возможной пренатальная диагностика с помощью УЗИ. Скоро станут доступными методы выявления мутаций гена COL2A1.
Лечение
Лечение хондродисплазий не разработано. Проводят симптоматическое лечение деформирующего остеоартроза и других проявлений болезни.
Многим больным проводят протезирование суставов и устранение расщелины неба.
Больных должен регулярно осматривать офтальмолог, чтобы своевременно обнаружить развитие катаракты и провести лазерную коагуляцию при отслойке сетчатки.
Больным советуют следить за весом и избегать контактных видов спорта.
При низкорослости очень важна психологическая помощь, с этой целью во многих странах создают группы поддержки.
Многие скелетные дисплазии сопровождаются различными аномалиями ребер, что приводит к уменьшению размеров грудной клетки, изменению ее формы, и, следовательно, - к вторичной гипоплазии легких. Изменения грудной клетки встречаются при танатофорной дисплазии, ахондрогенезе, несовершенном остеогенезе, кампомелической дисплазии, гипофосфатазии, которые были описаны выше, а также при асфиксической дисплазии грудной клетки (синдром Жене), синдроме коротких ребер - полидактилии, хондроэктодермальной дисплазии (синдром Эллиса - Ван-Кревельда), спондилоторакаль-ной дисплазии (синдром Ярхо-Левина) и некоторых других заболеваниях.
Поскольку форма и размеры грудной клетки принципиально влияют на состояние легких и, естественно, на перинатальные исходы, считаем необходимым представить описание наиболее часто встречающихся скелетных дисплазии, ведущими симптомами которых являются изменения грудной клетки.
Синдром Жене является крайне редким аутосомно-рецессивным состоянием встречающимся у 1:100 000-130 000 новорожденных.
Это заболевание характеризуется узкой колоко-лообразной грудной клеткой с короткими, толстыми, горизонтально расположенными ребрами, что приводит к возникновению гипоплазии легких и легочной недостаточности после рождения. Трубчатые кости могут быть слегка укорочены (по типу ризомелии) или не изменены вовсе. Укорочение конечностей, как правило, не определяется до 24 нед. Других изменений скелета в пренатальном периоде при этом синдроме нет.
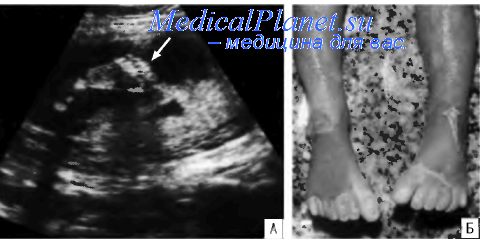
Синдром Жене может сопровождаться дисплазией почек, печени, поджелудочной железы, однако эти нарушения носят в основном морфологический характер и далеко не всегда подлежат пренатальной ультразвуковой диагностике.
Основным пренатальным диагностическим критерием является выраженное уменьшение размеров грудной клетки и изменение ее формы. В связи с редкостью этого заболевания и немногочисленностью эхографических пренатальных признаков в отечественной литературе нет описаний его дородовой диагностики.
Прогноз при синдроме Жене крайне вариабелен и зависит от степени поражения грудной клетки. Следует отметить, что строение легких при этом заболевании практически не изменено. Отмечается лишь уменьшение альвеолярного деления. Нарушение функции дыхания связано с резким ограничением дыхательных движений. Хирургическая коррекция этого порока не имеет хороших результатов.
При подозрении на синдром Жене необходимо очень тщательно изучить анамнез. Особого внимания заслуживают семьи с рождением ранее детей с ВПР ОДС, поскольку вероятность повторения этого заболевания составляет 25%.
Синдром коротких ребер - полидактилии (тип I, II, III) -летальная дисплазия с аутосомно-ре-цессивным типом наследования, характеризующаяся короткими ребрами, узкой грудной клеткой и постаксиальной полидактилией. По мнению исследователей, эта хондродистрофия возникает в результате нарушения клеточной дифференцировки на самых ранних этапахэмбриогенеза, поэтому она сопровождается аномалиями развития других органов и систем (лица, сердца, почек, желудочно-кишечного тракта).
Одним из ведущих симптомов этой хондродистрофии является резкое укорочение ребер, что приводит к нарушению формирования грудной клетки и вторичной легочной недостаточности. Полидактилия также характерна для этого синдрома, однако есть постнатальные описания клинических случаев, полностью соответствующих этому заболеванию, но не сопровождающихся увеличением количества пальцев.
Выделяют несколько типов этого заболевания. Тип I (Салдино - Нунан) помимо перечисленных признаков характеризуется узкими метафизами, тип II (Маевский) - наличием расщелин лица и диспропорционально укороченными большими берцовыми костями, тип III (Наумофф) - широкими метафизами, тип IV (Бимер - Лангер) - максимально укороченными ребрами, срединными расщелинами, а также наличием пупочной грыжи.
В целом ультразвуковая картина при этом синдроме достаточно богата. Помимо нарушения формирования ребер и соответственно изменений размеров и формы грудной клетки у плода обычно отмечаются множественные пороки развития. Нередко заболевание сопровождается изменением количества околоплодных вод и неиммунной водянкой. В связи с многочисленностью эхографических изменений в большинстве случаев в пренатальном периоде эта па-тология интерпретируется как множественные пороки развития плода.
В отечественной литературе опубликован единственный случай пренатальной диагностики синдрома коротких ребер - полидактилии. Клинический диагноз был основан на отягощенном анамнезе (прерывание беременности по медицинским показаниям по поводу наличия у плода хондродистрофии с полидактилией) и характерной ультразвуковой картине. Основную роль в постанове дородового диагноза сыграло обнаружение у плода постаксиальной полидактилии.

Эталон новых стандартов! Беспрецедентная четкость, разрешение, сверхбыстрая обработка данных, а также исчерпывающий набор современных ультразвуковых технологий для решения самых сложных задач диагностики.
Синдром выделен в отдельную нозологическую единицу R. Ellis и S. van Creveld в 1940 г. Синдром Ellis - van Creveld (хондроэктодермальная дисплазия, мезодермальная дисплазия, мезодермальная карликовость с шестипалостью) (MIM 225500) - редкое аутосомно-рецессивное состояние с переменными фенотипическими признаками, частота его составляет 1 случай на 60 000 живорождений 1. Синдром изучен достаточно хорошо, в литературе описаны десятки больных с такой патологией. Встречается он у представителей самых разных национальностей и расовых групп. Синдром наиболее распространен среди амишей (США), малочисленной религиозно-этнической общины потомков голландцев-протестантов с большой распространенностью близкородственных браков, что объяснимо, учитывая тип наследования данного заболевания 3.
Фенотипический спектр синдрома складывается из: 1) хондродисплазии, 2) полидактилии, 3) эктодермальной дисплазии, 4) пороков сердечно-сосудистой системы.
В названии "хондроэктодермальная дисплазия" отмечены главные особенности синдрома - аномалии хрящевого слоя костей между диафизом и эпифизом, а также дисплазия эктодермальной ткани (ногтей и зубов) [6].
Заболевание характеризуется симметричным укорочением конечностей легкой или средней степени (этот порок отмечен у всех больных), постаксиальной полидактилией, гипоплазией грудной клетки с укорочением ребер средней степени тяжести и пороками сердца (в 60% случаев), чаще всего представленными дефектами межпредсердной перегородки (ДМПП) [6, 7], иногда - единым предсердием, реже - дефектами межжелудочковой перегородки и клапанным стенозом аорты. Полидактилия является постоянным признаком заболевания. Дополнительный палец обычно имеет правильно сформированные пястные и фаланговые кости. Полидактилия на руках отмечается фактически в 80% случаев, на ногах - в существенно меньшем проценте случаев (10-25%). Из других пороков опорно-двигательной системы следует назвать косолапость (до 30% случаев) и вальгусное положение коленей.
Признаками эктодермальной дисплазии являются ногтевая дисплазия, гиподонтия, гипертрофия десен и запоздалое прорезывание зубов. У 70% новорожденных ногти кистей и стоп гипоплазированы, дистальные отделы обычно утолщены. В части случаев ногти могут отсутствовать. Рост волос на голове скудный, вплоть до обширных участков алопеции.
При синдроме Ellis - van Creveld детская летальность отмечается в 50% случаев. Смертность определена степенью выраженности гипоплазии грудной клетки, ведущей к вторичной гипоплазии легких, а также типом и выраженностью порока сердца. Выжившие дети обладают нормальным интеллектуальным развитием, но низким ростом. Окончательный рост от 109 до 162 см. Необходимость коррекции ДМПП не влияет на выживаемость или качество жизни, если преодолены дыхательные проблемы, связанные с грудной гипоплазией. Прочие признаки эктодермальной дисплазии легко поддаются терапии, хотя дефекты прорезывания зубов часто требуют проведения ортодонтических процедур [8]. Обычно нарушена функция кистей, в частности способность сжимать пальцы в кулак.
Пренатальная ультразвуковая диагностика синдрома Ellis - van Creveld потенциально возможна, начиная с середины II триместра беременности (табл. 1). Однако L. Dugoff и соавт. [4] сообщили об ультразвуковой диагностике синдрома в 12 нед.
| Авторы | Срок диагностики, нед |
|---|---|
| F. Cuillier [6] | 24 |
| N. Venkat-Raman и соавт. [9] | 18 |
| C. Sergi и соавт. [10] | 25 |
| T. Tongsong, P. Chanprapaph [11] | 27 |
| I. Torrente и соавт. [12] | 22 |
| F. Qureshi и соавт. [13] | 22-23 |
| О.A.R.M. Al-Asali [14] | 24 |
В отечественных журналах, посвященных проблемам пренатальной ультразвуковой диагностики, мы не встретили ни одного сообщения о дородовом выявлении данного синдрома. В связи с этим представляем собственный опыт пренатальной диагностики синдрома Ellis - van Creveld.
Беременная 35 лет, данная беременность первая. Мужу 37 лет, брак не родственный, производственных вредностей супруги не имеют. На учете в женской консультации пациентка состояла с 17 нед беременности. Ультразвуковое исследование проводилось неоднократно (в сроки 17, 23, 28, 32 нед). В сроки 17 и 23 нед беременности в проколах ультразвуковых обследований указано на отсутствие патологии у плода, в срок 28 нед отмечено уменьшение длины трубчатых костей, поставлен диагноз гипохондроплазии. На ультразвуковое исследование 2-го уровня женщина не была направлена. В срок 32 нед повторно отмечено укорочение конечностей, повторно поставлен диагноз гипохондроплазии.
Впервые в медико-генетическое отделение МОНИИАГ женщина обратилась при сроке беременности 37 нед. Ультразвуковое обследование проводилось на сканере Medison Accuvix-XQ с использованием режима поверхностной объемной реконструкции 3D/4D.
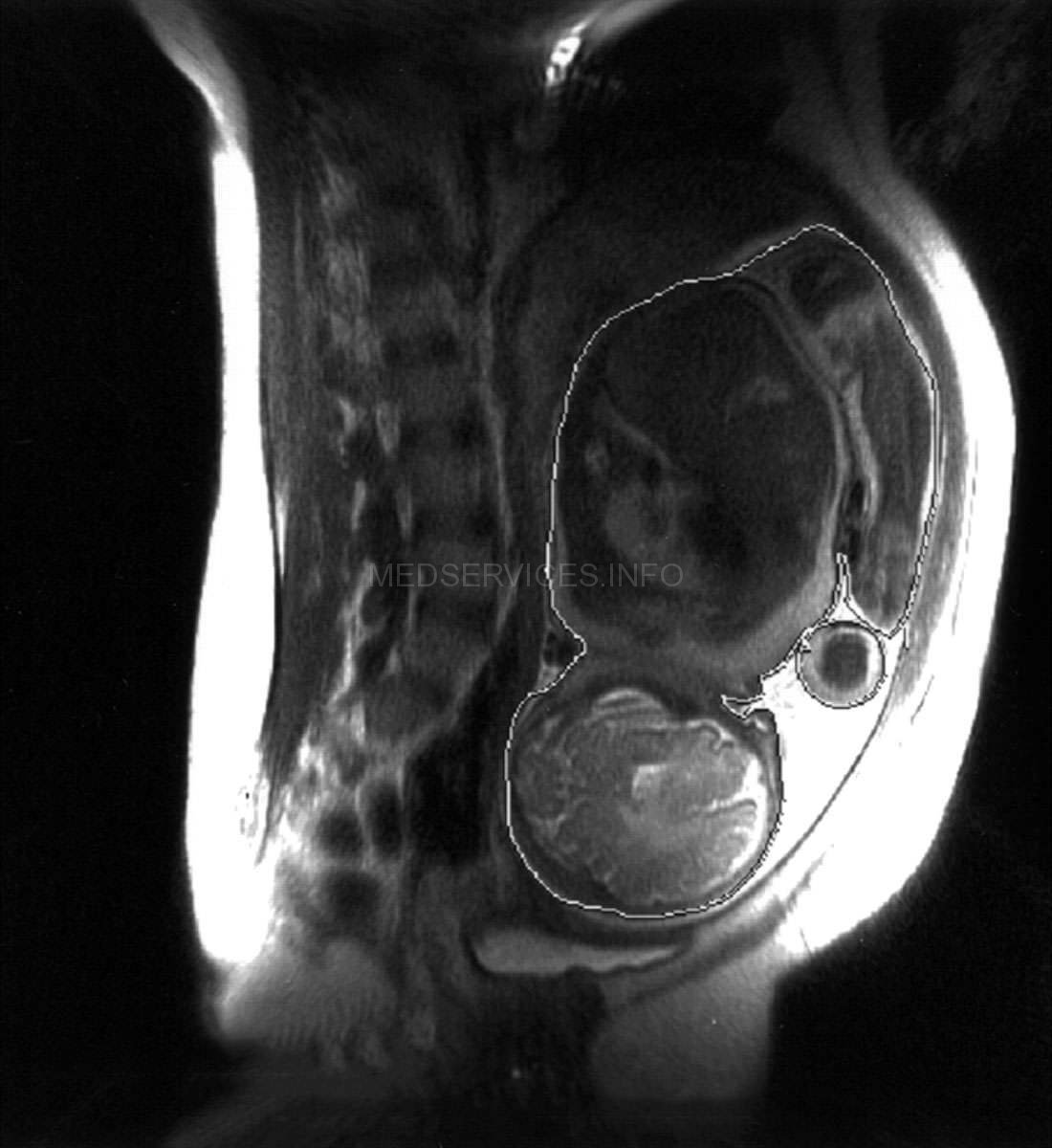
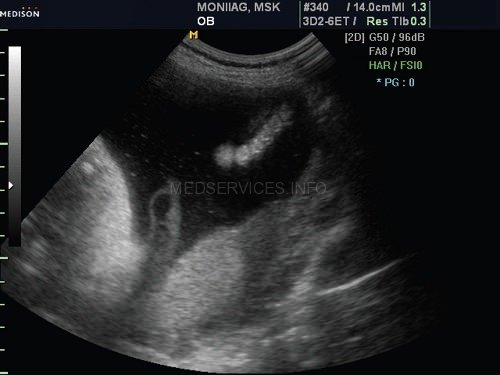
При эхографии в 37 нед беременности на фоне многоводия был обнаружен один живой плод мужского пола с аномальным внешним видом. Все длинные трубчатые кости были укорочены и соответствовали 26 нед гестации (рис. 1).
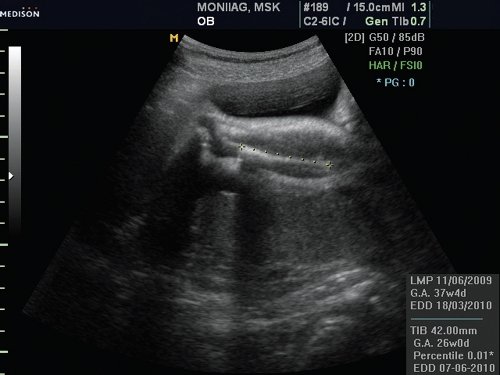
Рис. 1. Укорочение костей голени (микромелия).
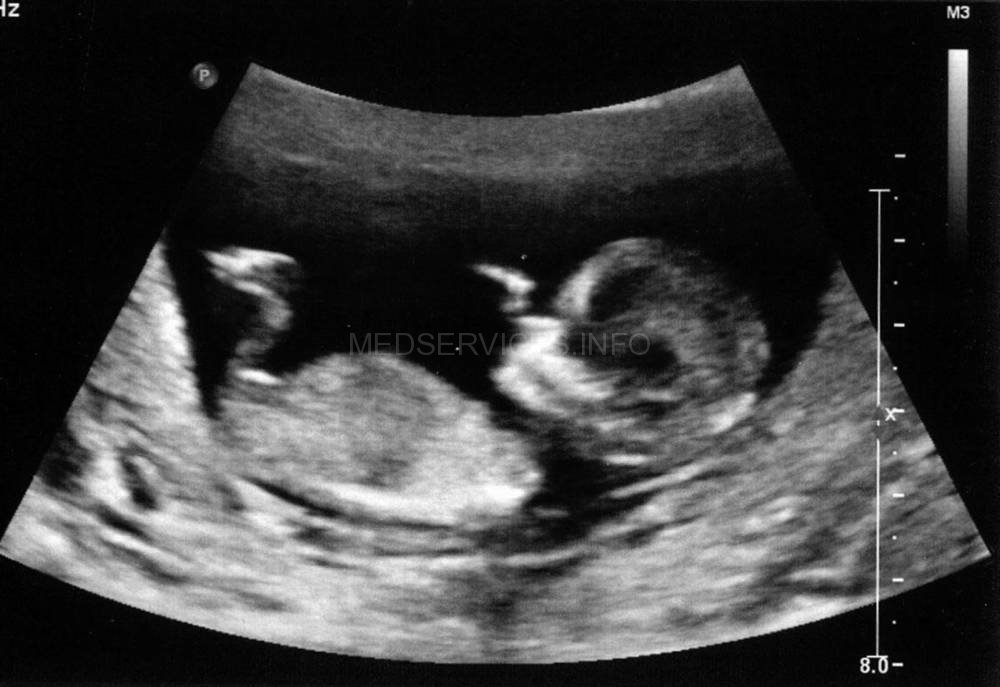
Грудная клетка была гипоплазирована. Ее размеры резко диссоциировали с размерами живота плода (рис. 2, 3).




- Здоровье
- Статьи
- Медицина
- Форум
- Rating
- Full Article
- Comments

Хондроэктодермальная дисплазия — проявляется после 25 нед. беременности в виде комплекса признаков: незначительное укорочение конечностей, полидактилия кистей и иногда порок сердца. Биометрические показатели головы и туловища находятся в пределах нормальных значений. Подкожная жировая клетчатка развита обычно. Порок совместим с внеутробной жизнью, хотя в некоторых случаях сопровождается нарушениями интеллектуального развития.
Синдром коротких ребер — полидактилия имеет два типа клинических проявлений. Общие признаки заболевания: выраженное укорочение конечностей, умеренная пренатальная гипоплазия, узкая грудная клетка, полидактилия кистей или стоп. Для I типа обличительными признаками служат: расщелина верхней губы и неба, часто поликистоз почек, иногда пороки сердца. При II типе чаще всего встречаются пороки пищеварительного тракта, косолапость. Искривление длинных трубчатых костей носит слабовыраженный характер. Сроки ультразвуковой идентификации данного заболевания приходятся на 20-25 нед. беременности. Прогноз неблагоприятный, так как новорожденные почти всегда погибают из-за выраженной гипоплазии легких и сопутствующих пороков.
Несовершенный остеогенез — врожденное заболевание, проявляющееся нарушением процесса минерализации костей плода. Выделяют IV типа заболевания, различающиеся по клиническим проявлениям. Возможна ультразвуковая диагностика только II и III типа. I и IV типы в антенатальном периоде эхографических проявлений не имеют. Сроки манифестации ультразвуковых признаков несовершенного остеогенеза II и Ш типа приходятся на 22-25 нед. беременности.
При эхографическом исследовании II тип характеризуется низкой минерализацией когтей скелета, включая кости черепа и тел позвонков.
Длинные трубчатые кости имеют множественные переломы, что приводит к деформации и некоторому укорочению.
В некоторых случаях удается визуализировать ложные суставы, характеризующиеся явным дафектом целостности кости. Вследствие тех же причин отмечают уменьшение грудной клетки. Голова плода при надавливании датчиком поддается видимой деформации, исчезающей после уменьшения давления. Данный тип абсолютно несовместим с внеутробной жизнью. Большинство плодов погибают в антенатальном периоде или в родах.
Эхографическая диагностика III типа несовершенного остеогенеза возможна, если удается выявить единичные переломы длинных трубчатых костей.
В родах количество переломов резко возрастает, что может приводить к гибели ребенка от болевого шока. Иногда такие переломы ошибочно рассматривают как последствия грубого ведения родового акта. Прогноз для внеутробной жизни плохой, хотя известны случаи жизни до 40-50 лет. Как правило, это тяжелые инвалиды, постоянно пребывающие в ортопедических стационарах, с рецидивирующими переломами.
Гипофосфатазия — редкое заболевание, также сопровождающееся нарушением минерализации костей. Эхографически характеризуется снижением эхогенности костей и уменьшением их толщины. Данные критерии достаточно сложны для объективной диагностики и требуют большого клинического опыта врача ультразвуковой диагностики. Биометрические параметры головы и конечностей находятся в пределах нормальных значений Кости свода черепа могут иметь очаги деминерализации, которые проявляются зонами повышенной проходимости ультразвука через голову плода.
Артрогрипоз — врожденное заболевание опорно-двигательного аппарата плода. В основе его лежит поражение задних рогов спинного мозга с последующим нарушением иннервации мышц конечностей и развитием их атрофии. Следствие этого — уменьшение мышечной массы пораженных конечностей и отсутствие движений в крупных суставах.
Эхографическое изображение артрогрипоза в первую очередь характеризуется истончением конечностей, их неподвижностью в коленных, голеностопных и лучезапястных суставах. На фоне тонких конечностей суставы выглядят утолщенными.
Коленные суставы чаще всего находятся в разогнутом положении.
Для голеностопных и лучезапястных суставов характерна эквиновальгусная деформация. Биометрические параметры костей плода при артрогринозе соответствуют сроку беременности.
Минимальные сроки обнаружения указанных изменений приходятся на 15-25 нед. беременности.
Артрогрипоз не является летальным пороком. Он поддается ортопедической коррекции, однако требует длительного пребывания ребенка в специальных лечебных учреждениях. Вопрос о тактике ведения беременности необходимо решать с учетом мнения родителей, которых обязательно следует уведомить о характере заболевания будущего ребенка. При этом следует помнить, что в некоторых случаях артрогрипоз может стать одним из проявлений более серьезных поражений ЦНС пледа.
Изолированные пороки развития конечностей плода — одна из трудных диагностических проблем. Основная проблема заключается в том, что визуализация всех отделов конечностей в некоторых случаях не представляется возможной. Социальная значимость данных пороков состоит в том, что большинство из них совместимы с внеутробной жизнью, а в некоторых случаях ведут к инвалидности ребенка.
С целью снижения числа диагностических ошибок при УЗИ необходимо исследование всех костей конечностей в продольном и поперечном сечении.
В структуре изолированных пороков конечностей одно из ведущих мест принадлежит дизмелическим поражениям конечностей. В основе этих пороков лежит тотальная или частичная аплазия определенных трубчатых костей. В зависимости от локализации и характера поражения принято выделять следующие формы дизмелий: проксимальную, аксиальную и дистальную эктромелии. Для проксимальной дизмелии характерно недоразвитие верхних участков длинных трубчатых костей. Минимальные сроки диагностики указанных пороков обычно составляют 15-20 нед. беременности.
Аксиальную дизмелию встречают редко. Данное заболевание характеризуется поражением как проксимальных, так и дистальных отделов конечностей. Недоразвитие всегда носит частичный характер. Порок также выявляют в сроки 18-25 нед. беременности.
При тибиальном типе порок проявляется недоразвитием большеберцовой кости разной степени выраженности. Всегда сопровождается выраженной деформацией голеностопного сустава.
Читайте также:
