
Синдрома верднига хоффмана мышечной атрофии

Генетические заболевания поражающие нервную систему приводят к поражению органов и частей организма, нарушают их нормальное функционирование. К одним из них и относится болезнь Верднига-Хоффмана. Встречается достаточно редко – один случай на 7-10 тысяч человек.
Этиология заболевания Верднига-Хоффмана
Болезнь Верднига-Хоффмана (спинальная мышечная амиотрофия) характеризуется патологией нервных клеток спинного мозга, следствием чего является усыхание мышечных волокон, переплетающихся со здоровыми. Этот процесс обусловлен недостаточным количеством протеина, отвечающего за выживаемость двигательных нейтронов. Существуют формы заболевания не связанные с данной патологией, вызванные другими модифицирующими факторами.
Нарушение работы нервных клеток приводит к разрастанию соединительной ткани, которая замещает мышечную. У пациента нарушается процесс глотания, опорно-двигательная и дыхательная функции. Психическое развитие не затрагивается. Чувствительность затронутых болезнью частей тела не снижается.
Заболевание Верднига-Хоффмана наследственное, передается от двух родителей, которые являются носителями патологического гена SMN, расположенного в 5-ой хромосоме. При этом симптомы болезни у них отсутствуют. У такой пары могут рождаться здоровые дети либо также носители гена, вероятность появления на свет больного малыша 25%.
Известные люди имеющие это заболевание: английский ученый-астрофизик Стивен Хокинг и российский IT специалист Валерий Спиридонов из Владимира.
Симптомы болезни
Признаки заболевания напрямую зависят от ее формы, при исследовании выявляются следующие клинические показатели:
- нарушение питания мышечных клеток приводит к их гибели. Сначала поражаются мышцы туловища, в первую очередь, спины, затем процесс переходит в зону плеч, бедер, конечностей;
- нарастающие болевые ощущения;
- снижение мышечного тонуса;
- подергивание мышц;
- уменьшение диаметра длинных костей, выявляемое посредством рентгенограммы;
- искривление позвоночника в одну сторону и назад;
- устоявшееся ограничение работы мышц (не сгибается и не расслабляется).
Симптомы, указывающие на наличие спинальной мышечной амиотрофии:
- слабость мышц, проявляется в нарушении двигательных процессов;
- за счет истончения кости, уменьшаются конечности;
- скудость мимических движений;
- глотательные и сосательные рефлексы снижены либо отсутствуют;
- при поражении межреберных мышц – нарушение дыхания, и как следствие воспалительные и застойные процессы в бронхах и легких;
- деформация скелетного аппарата в зоне грудной клетки и позвоночника;
- тремор рук и ног;
- торможение процессов физического развития.
Формы и стадии заболевания
Спинальная мышечная амиотрофия в большинстве случаев проявляется в первый год жизни ребенка. Чем раньше, тем более тяжелое ее течение. Уровень смертности высок, в основном дети умирают до 4 лет, редко – до 20. Может проявиться и у взрослых людей. Различают три основных формы заболевания:
- Врожденная болезнь Верднига-Хоффмана. Первые симптомы появляются сразу после рождения либо еще во время внутриутробного периода. При этом движения плода затихают. У новорожденного наблюдается нарушение процессов дыхания, сосания и глотания. Ребенок не держит голову, не переворачивается, слабо кричит. Течение болезни тяжелое, острое, продолжительность жизни коротка, до 2 – 2,5 лет. Однако в некоторых случаях с помощью современных аппаратов искусственной вентиляции легких и кормление не через зонд, а непосредственно в желудок, жизнь пациента можно продлить. Умственно и эмоционально ребенок развивается без нарушений.
- Вторая форма, ранняя детская. Развитие ребенка протекает в соответствии нормам. Он начинает вовремя держать головку, переворачиваться. До полугода родители не замечают никаких симптомов. После перенесенной инфекции болезнь проявляется в виде периферийных параличей сначала нижних, затем верхних конечностей, в конечном итоге – всего туловища, приобретенные навыки утрачиваются, тонус мышц снижается. Возникает тремор пальцев рук, непроизвольные мышечные сокращения языка. На более поздней стадии присоединяется затрудненная работа дыхательной системы. Течение болезни не стремительное, как при врожденной форме, некоторые дети могут доживать до подросткового возраста. Прогноз заболевания зависит от степени поражения мышц, отвечающих за дыхательный процесс.
- Третья форма, поздняя. Первые симптомы проявляются после 2 лет. К этому времени малыш уже развит физически и психологически согласно возрастным нормам. Прогрессирование заболевания происходит медленно, постепенно, характеризуется вялостью и неуклюжестью ребенка при ходьбе и других двигательных процессах. Развивается парез конечностей, угасание глотательного и сухожильного рефлекса, признаки бульбарного паралича, а также деформация костной ткани. Третья форма протекает мягче первых двух, пациенты могут прожить до 30 лет.
Выделяют формы спинальной мышечной амиотрофии, проявляющиеся в более позднем возрасте.
- Болезнь Кульдберга-Веландера считается наиболее легкой формой атрофии детского возраста. В большинстве случаев, начало болезни приходит на подростковоый период, но бывают и более ранние проявления.
Известны случаи, когда пациенты не утрачивают способности ходить и обслуживать себя самостоятельно, проживая долгую жизнь.
- Болезнь Кеннеди связанна с мутацией гена в Х-хромосоме, передается девочкам от двух родителей, мальчикам от мамы. Проявляется во взрослом возрасте.
Злокачественное течение врожденной формы Верднига-Хоффмана, дает мало шансов для планирования будущего таких детей, однако при 2 и 3 форме можно продлить жизнь ребенку, важно вовремя реагировать на инфекционные заболевания, которые резко ухудшают состояние больного и приводят к появлению новых симптомов, наихудший из которых – нарушение дыхательной функции.
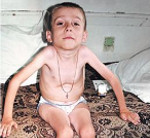
Внешние проявления болезни Верднига-Хоффмана
 |
В чем заключается опасность заболевания?
В связи с тем, что болезнь Верднига-Хоффмана неизлечима, самой главной опасностью является летальный исход. При врожденной форме дети живут достаточно малый период времени, заболевание быстро прогрессирует и не оставляет шансов на выживание.
С помощью современных исследований, существует возможность в период беременности выявить наличие заболевания у плода и не допустить рождения тяжело больного ребенка.
В других формах болезнь проявляет первые признаки после перенесенной кишечной или распираторной инфекции, в дальнейшем родители под руководством лечащих врачей ограничивают возможность развития инфекции у ребенка, которая усугубит ее течение и несет смертельную опасность. Однако бронхиты, пневмонии и другие заболевания лор-органов часто встречаются у болеющих заболеванием Верднига-Хоффмана.
Диагностика и лечение болезни Верднига-Хофмана
На ранних стадиях заболевания бывает трудно дифференциировать заболевание, так как симптоматика может быть схожа с другими болезнями:
- острый полиомиелит отличается отсутствием прогрессирования заболевания и несимметричными параличами;
- миопатия – также имеет наследственное происхождение, имеет прогрессивное течение, но причиной слабости мышц является нарушение в них обменных процессов;
- врожденная миатония наиболее схожа с болезнью Верднига-Хоффмана, отличить их достаточно можно с помощью биопсии мышечной ткани.
Для диагностики заболевания неврологу понадобятся данные о первом проявлении симптомов, характере их развития, наличия сопутствующих заболеваний.
Проводится ряд исследований для постановки диагноза:
- Электронейромиография выявляет нарушения в работе нервно-мышечной системы. Наблюдаются изменения по мышечному типу, что указывает на патологию двигательного нейтрона;
- Генетический анализ выявляет мутацию гена SMN;
- Биохимия крови на уровень креатинкиназы, показатели в пределах нормы не исключают заболевание;
- Биопсия мышц для морфологического исследования, которое выявляет пучковую атрофию мышечных волокон, чередующихся со здоровыми, а также разрастание соединительной ткани;
- МРТ для исключения других заболеваний.
Для проведения диагностики плода внутриутробно применяется метод биопсии хориона, кордоцентез, амниоцентез. Выявление заболевания является показанием к прерыванию беременности. Вылечить пациента с болезнью Верднига-Хоффмана невозможно. Для продления жизни и улучшения ее качества применяют симптоматическое лечение. Развитие болезни и ухудшение симптомов сдерживают путем обеспечения работы обменных процессов в мышечной ткани.
С помощью лечебной физкультуры и массажа улучшается кровообращение, снижается риск застоев, поддерживается работоспособность мышц, предотвращается неподвижность суставов и потеря ими эластичности. Нагрузки должны быть непродолжительными и осторожными. Физиотерапия способствует удержанию двигательных навыков на имеющемся уровне, их укрепление. Специальные приспособления помогут самостоятельно передвигаться, пользоваться компьютером и даже писать. Портативные аппараты ИВЛ дают возможность пациентам находиться за пределами стационара и проживать жизнь продуктивнее.
Прогноз болезни Верднига-Хоффмана
Прогноз для данного заболевания достаточно неблагоприятен. Шансов на выздоровление нет. Единственным способом продления жизни является своевременное лечение, здоровое питание и разумные физические нагрузки. Дети с врожденной формой Верднига-Хоффмана погибают в пределах 6 месяцев - 2 лет. Более позднее проявление болезни дает больше времени для жизни.

Амиатрофия Верднига-Гоффмана относится к орфанным (редким) патологиям, частота встречаемости которой 1:6000. Характеризируется крайне тяжелым течением и быстрым прогрессированием, приводящим к стойким и выраженным деформациям опорно-двигательного аппарата и летальному исходу.
- Что это?
- Причины заболевания
- I тип амиотрофии
- II тип
- III тип
- IV тип
- Диагностика
- Методы лечения
- Прогноз: сколько живут пациенты?
- Что нужно запомнить?
Что это?
Спинальная амиотрофия – генетическое заболевание, при котором обнаруживается мутации в 5 хромосоме в генах SMN1 и SMN2. Эти гены отвечают за сохранение и нормальное функционирование мотонейронов – нервов, которые отправляют импульс к скелетной мускулатуре. В результате отсутствия стимуляции мышц они атрофируются. У больных отмечается дегенеративные процессы в спинном мозге: разрушаются двигательные нейроны, нарушается функционирование передних рогов спинного мозга. В головном мозге изменения происходят в двигательных ядрах.
При мышечной амиотрофии Верднига мутирует ген SMN1, который препятствует гибели мотонейронов. Ген SMN2 выполняет эту функцию только частично и его возможности быстро истощаются. Течение и тяжесть патологии зависит от локализации и объема поражения в генном аппарате.

Фото 2
Спинальная амиотрофия вне зависимости от ее типа и формы – генетическое врожденное заболевание. Зачастую, первые признаки патологии обнаруживаются в младенческом возрасте либо еще во время беременности матери ребенка.
Одинаково распространена как у мужчин, так и женщин. Основное условие ее развития – наличие дефектного гена у обоих родителей, которые могут быть здоровыми и являются лишь носителями дефектной хромосомы.
В результате нарушения проводимости нервных импульсов от мотонейронов к мускулатуре происходит атрофия мышц нижних и верхних конечностей, диафрагмы, органов ЖКТ, сердца и другие. На фоне этого деформируются кости и суставы. Наиболее опасным проявлением является нарушения дыхательной функции и работы сердца.
В зависимости от тяжести течения, локализации дефектов и клинических проявлений выделяют 4 формы амиотрофии. Пациенты с любой формой амиотрофии являются инвалидами. Они не способны самостоятельно себя обслуживать, и нуждаются в постоянном уходе и медицинском наблюдении.
Причины заболевания
Основная причина развития спинальной амиотрофии – генетическая мутация у родителей и передача рецессивного гена плоду. Но, что привело к ее возникновению, ученым еще не известно. Мутация может произойти спонтанно, без видимых на то причин. Предполагаемыми причинами болезни являются следующие факторы:
- вирусные заболевания;
- нарушение экологии;
- прием во время беременности наркотиков, алкоголя, курение;
- воздействие терратогенных факторов;
- ионизирующее излучение.
В большинстве случаев родители узнают о том, что являются носителями опасного гена, только после рождения ребенка с диагнозом.
При некоторых вариантах амиотрофии толчком для прогрессирования мышечной дистрофии могут быть перенесенные вирусные заболевания, инсоляция, гормональный сбой.
I тип амиотрофии
Наиболее злокачественный и распространенный тип патологии – амиотрофия Верднига-Гоффмана. Диагностируется у детей раннего возраста либо внутриутробно. У детей после рождения отмечается снижение и угасание всех рефлексов. Им сложно сосать грудь, из-за чего часто возникает необходимость кормить ребенка через зонд.
Мышечная гипотония приводит к слабости шейных мышц – дети не могут научиться держать головку, самостоятельно переворачиваться, сидеть, стоять и ходить. У большинства детей возникают трудности с глотанием. В большинстве случаев амиотрофию диагностируют в течение 6 месяцев после рождения. Родители обращают внимание на вялость ребенка и малоподвижность, прогрессирующее снижение мышечного тонуса. Также младенцы плохо набирают вес.
Характеризируется поражением диафрагмы, которая участвует в дыхательном акте. Многие пациенты самостоятельно не дышат. Из-за выраженной дыхательной недостаточности многие дети не доживают до года. Продлить им жизнь удается с помощью ИВЛ и питания через зонд. 95% детей умирают до 4 лет.
Патология сопровождается прогрессирующими деформациями опорно-двигательной системы.
Помимо поражения мышц и костей, часто выявляется отставание в умственном развитии.
Справка. Заподозрить диагноз можно еще во время беременности матери, когда отмечается слабое шевеление плода, нарушение сердечной деятельности, отставание в развитии.
II тип
Промежуточная амиотрофия. Диагностируется у детей 3 месяцев—1,5 года. В более раннем возрасте диагноз трудно установить из-за особенностей мышечной системы у детей. Из-за выраженной мышечной гипотонии и снижения сухожильных рефлексов младенцы не могут научиться ползать и ходить. Лишь в 25% возможно, что ребенок сможет самостоятельно сесть.
Сопровождается поражением костно-суставной системы. У многих искривляется позвоночник, деформируется грудная клетка, атрофируются суставы рук и ног. Родители отмечают похудение ребенка либо прекращение набора веса.
Особенность этого типа амиотрофии – возможное внезапное прекращение ее развития. Несмотря на ремиссию, утратившиеся способности практически невозможно восстановить. Прогрессирование болезни может возобновиться либо ускориться после перенесенного ОРВИ или ОРЗ и других заболеваний, снижающих иммунитет. Из-за атрофии диафрагмы нарушается работа легких, ослабевает работа легких, из-за чего возникает одышка и тахикардия. Вовлечение в процесс мускулатуры органов пищеварения, бульбарные нарушения приводят к неспособности самостоятельно принимать пищу.
III тип
Болезнь Кугельберга-Веландера – вариант поздней амиотрофии. При данной форме диагноза первые признаки возникают после двух лет, либо во взрослом возрасте. Дети, которые уже умеют ходить, внезапно становятся неловкими, жалуются на боль в ногах при хождении. Они слишком часто падают, неуверенно ходят и практически перестают бегать. Многие родители замечают изменения походки и внезапно появившуюся неуклюжесть. Вначале развития патологии поражаются мышцы ног, затем атрофируются мышцы верхних конечностей и других частей тела. Из-за снижения объема мышц также отмечается снижение веса.
У некоторых больных может наступить период ремиссии, в который прекращается прогрессирование диагноза. Этот период может продлиться от нескольких недель, до нескольких десятков лет.
Нарушение дыхательной функции наиболее опасное проявление атрофии мускулатуры.
Со стороны психики и умственного развития нарушения не выявляются.
IV тип
Первые признаки и симптомы появляются в 30-50 лет. Основными жалобами является слабость мышц, тремор. Из-за атрофии появляются контрактуры в области суставов, с ограничением их подвижности. Больные резко худеют.
Часто сопровождается искривлением позвоночника и деформацией грудной клетки.
Первыми в процесс вовлекаются мышцы ног, после постепенно вовлекаются руки. Как правило, дыхательная и глотательная функция не нарушена.
Большинство пациентов самостоятельно ходят, но хромают, испытывают боль и дискомфорт. В редких случаях прогрессирование амиотрофии приводит к потере способности ходить, и больные вынуждены передвигаться на инвалидной коляске.
Диагностика
Основным методом диагностики является генетическое исследование. Для проведения тестирования используют кровь. В некоторых случаях для подтверждения диагноза проводят биопсию пораженных тканей.
Для определения тяжести болезни, объема поражений и нарушения функционирования мышечной системы проводят электромиографию.
Также обследовать необходимо и других близких родственников пациента.
Важно! Для пренатальной диагностики исследуют околоплодные воды. Такой метод применяется только при наличии симптомов и генетической предрасположенности у родителей.
Методы лечения
Генетическое заболевание является неизлечимым и практически не поддается коррекции. При прогрессирующем течении практически невозможно предотвратить и остановить мышечную атрофию. Несмотря на развитие генной инженерии, ученым не удалось создать лекарство, способное устранить генетический сбой.
Лечение направлено на поддерживание жизненно важных функций организма, облегчение состояния больного и предотвращение деформации скелета. Для этого применяют следующие методы:
- искусственная вентиляция легких – показана при дыхательной недостаточности;
- физиопроцедуры – массаж, ЛФК, электрофорез, лечебные ванны и другие;
- ношение корсета и других ортопедических приспособлений;
- витаминотерапия;
- применение ноотропов и препаратов, улучшающих питание нервной ткани: актовегин, трентал, церебролизин и другие.
Паллиативная терапия направлена на облегчение состояние тяжелобольных пациентов.
Ученые всего мира пытаются создать препарат для лечения патологии. Известно, что уже разработано несколько препаратов, которые возможно сумеют помочь пациентам. Они находятся на стадии клинических испытаний и их эффективность и безопасность для организма только изучается.
Родителям детей с тяжелым, неподдающимся лечению, диагнозом, для рождения здорового ребенка необходимо планировать беременность с репродуктологом. Искусственное оплодотворение, использование донорской спермы или яйцеклетки, экстракорпоральное оплодотворение – одни из способов родить здорового ребенка носителям дефектного гена.
Прогноз: сколько живут пациенты?
Спинальная амиотрофия крайне тяжелое заболевание, которое существенно ухудшает качество жизни больных и является одной из причин младенческой смерти. Пациенты с таким диагнозом являются инвалидами и зачастую не в силах самостоятельно себя обслуживать.
Многие пациенты не могут ходить и стоять, передвигаясь с помощью инвалидной коляски. При поражении рук больные не способны делать элементарных вещей: есть, держать небольшие предметы, читать, умываться и прочее. Они нуждаются в дополнительном уходе и постоянном медицинском наблюдении. При тяжелом течении они дышать лишь с помощью аппарата ИВЛ и питаются через зонд.
Продолжительность жизни во многом зависит от формы заболевания. Наиболее тяжелой является амиотрофия Верднига, при которой больные дети редко достигают 4 лет. Пациенты со вторым типом болезни редко доживают до совершеннолетия. Наиболее благополучным являются второй и третий тип болезни, при которых пациенты достигают зрелого возраста.
В настоящее время спинальная амиотрофия относится к неизлечимым заболеваниям, часто приводит к летальному исходу.
Амиотрофия Верднига-Гоффмана — это наиболее злокачественная спинальная мышечная атрофия, развивающаяся с рождения или в первые 1-1,5 года жизни ребенка. Характеризуется нарастающими диффузными мышечными атрофиями, сопровождающимися вялыми парезами, прогрессирующими до полной плегии. Как правило, амиотрофия Верднига-Гоффмана сочетается с костными деформациями и врожденными аномалиями развития. Диагностическую основу составляет анамнез, неврологический осмотр, электрофизиологические и томографические исследования, анализ ДНК и изучение морфологического строения мышечной ткани. Лечение слабо эффективно, направлено на оптимизацию трофики нервной и мышечной тканей.
МКБ-10
- Причины
- Патогенез
- Симптомы амиотрофии
- Врожденная форма
- Ранняя детская форма
- Амиотрофия Кугельберга-Веландера
- Диагностика
- Лечение амиотрофии Верднига-Гоффмана
- Немедикаментозная терапия
- Медикаментозная терапия
- Хирургическое лечение
- Новейшие разработки
- Прогноз
- Профилактика
- Цены на лечение
Общие сведения
Амиотрофия Верднига-Гоффмана является самым тяжелым вариантом из всех спинальных мышечных атрофий (СМА). Ее распространенность находится на уровне 1 случай на 6-10 тыс. новорожденных. Заболевание имеет несколько форм: врожденную, промежуточную (раннюю детскую) и позднюю. Целый ряд специалистов выделяет последнюю форму как самостоятельную нозологию — амиотрофию Кугельберга-Веландера. Отсутствие этиотропного и патогенетического лечения, ранний летальный исход ставят курирование пациентов с болезнью Верднига-Гоффмана в ряд наиболее сложных задач, стоящих перед современной неврологией и педиатрией.
Причины
Амиотрофия Верднига-Гоффмана — наследственная патология, кодируемая поломкой в генетическом аппарате на уровне локуса 5q13 5-й хромосомы. Ген, в котором происходят мутации, получил название survival motor neuron gene (SMN) — ген, ответственный за выживание мотонейронов. У 95% пациентов с болезнью Верднига-Гоффмана отмечается делеция теломерной копии этого гена. Тяжесть СМА прямо коррелирует с протяженностью участка делеции и сопутствующим наличием изменений (рекомбинации) в генах H4F5, NAIP, GTF2H2.
Носителем измененного гена, обуславливающего возникновение заболевания, является каждый 50-й человек. Но благодаря аутосомно-рецессивному типу наследования, патология у ребенка проявляется только тогда, когда соответствующая генетическая аберрация имеется и у матери, и у отца. Вероятность рождения ребенка с патологией в такой ситуации составляет 25%.
Патогенез
Результатом аберрации SMN-гена является недоразвитие мотонейронов спинного мозга, локализующихся в его передних рогах. Следствием становится недостаточная иннервация мышц, приводящая к их выраженной атрофии с потерей мышечной силы и прогрессирующим угасанием способности совершать активные двигательные акты. Основную опасность представляет слабость мышц грудной клетки, без участия которых невозможны движения, обеспечивающие дыхательную функцию. При этом сенсорная сфера на всем протяжении заболевания остается интактной.
Симптомы амиотрофии
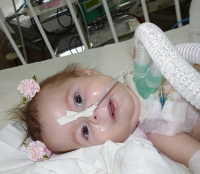
СМА I клинически манифестирует до 6-месячного возраста. Внутриутробно может проявляться вялым шевелением плода. Зачастую мышечная гипотония отмечается с первых дней жизни и сопровождается угасанием глубоких рефлексов. Дети слабо кричат, плохо сосут, не могут держать голову. В отдельных случаях (при более позднем дебюте симптомов) ребенок учится держать голову и даже сидеть, но на фоне развития заболевания эти навыки быстро исчезают. Характерны ранние бульбарные нарушения, понижение глоточного рефлекса, фасцикулярные подергивания языка.
Данная амиотрофия Верднига-Гоффмана сочетается с олигофренией и нарушениями формирования костно-суставного аппарата: деформациями грудной клетки (воронкообразной и килевидной грудной клеткой), искривлением позвоночника (сколиозом), контрактурами суставов. У многих пациентов выявляются другие врожденные аномалии: гемангиомы, гидроцефалия, косолапость, дисплазия тазобедренных суставов, крипторхизм и пр.
Течение СМА I наиболее злокачественное с быстро нарастающей обездвиженностью и парезом дыхательной мускулатуры. Последний обуславливает развитие и прогрессирование дыхательной недостаточности, выступающей основной причиной летального исхода. В связи с нарушением глотания возможен заброс пищи в дыхательные пути с развитием аспирационной пневмонии, которая может явиться смертельно опасным осложнением спинальной амиотрофии.
СМА II дебютирует после 6-месячного возраста. К этому периоду дети имеют удовлетворительное физическое и нервно-психическое развитие, в соответствии с возрастными нормами приобретают навыки держать голову, переворачиваться, садиться, стоять. Но в подавляющем большинстве клинических случаев дети так и не успевают научиться ходить. Обычно эта амиотрофия Верднига-Гоффмана манифестирует после перенесенной ребенком пищевой токсикоинфекции или другого острого инфекционного заболевания.
В начальном периоде периферические парезы возникают в нижних конечностях. Затем они достаточно быстро распространяются на верхние конечности и мускулатуру туловища. Развивается диффузная мышечная гипотония, происходит угасание глубоких рефлексов. Наблюдаются контрактуры сухожилий, тремор пальцев, непроизвольные мышечные сокращения (фасцикуляции) языка. На поздних стадиях присоединяются бульбарные симптомы, прогрессирующая дыхательная недостаточность. Течение более медленное, чем у врожденной формы болезни Верднига-Гоффмана. Пациенты могут доживать до 15-летнего возраста.
СМА III - наиболее доброкачественная спинальная амиотрофия детского возраста. Манифестирует после 2-х лет, в отдельных случаях в период от 15 до 30 лет. Отсутствует задержка психического развития, длительное время пациенты способны самостоятельно двигаться. Некоторые из них доживают до глубокой старости, не теряя способности к самообслуживанию.
Диагностика
Больные спинальной мышечной атрофией I типа находятся под наблюдением детских неврологов и неонатологов. Большое значение имеет возраст манифестации заболевания – для амиотрофии Верднига-Гоффмана характерно развитие с момента рождения до 6 месяцев. В анамнезе часто имеются сведения о позднем и вялом шевелении плода во время беременности.
- Лабораторные тесты. В биохимическом анализе крови обнаруживается небольшое повышение концентрации креатинфосфокиназы. У некоторых пациентов данный показатель может находиться в пределах нормальных значений. При анализе газов крови выявляется снижение парциального давления кислорода (paO2) и увеличение углекислого газа (paCO2).
- Спирометрия. Вследствие выраженной мышечной слабости дыхательной мускулатуры при измерении функции внешнего дыхания отмечаются рестриктивные нарушения в виде снижения жизненной емкости легких.
- ЭНМГ. При выполнении игольчатой электронейромиографии удается зафиксировать следующие изменения – резкое уменьшение скорости проведения и амплитуды вызванных потенциалов, спонтанную биоэлектрическую активность в покое (фасцикуляции, фибрилляции).
- Гистология. При патоморфологическом исследовании мышечного биоптата обнаруживается пучковая атрофия мышечных волокон, чередующаяся с неизмененной мышечной тканью, гипертрофированными миофибриллами и соединительнотканными разрастаниями.
- ДНК-анализ. Верифицирующий тест, позволяющий достоверно установить диагноз. Методом полимеразной цепной реакции выявляется генетическая мутация (делеция) SMN1 экзона 7.
Амиотрофию Верднига-Гоффмана следует дифференцировать с другими генетически обусловленными нервно-мышечными заболеваниями, имеющими такое же быстропрогрессирующее течение. К ним относятся врожденные структурные миопатии, ювенильный боковой амиотрофический склероз, синдром Фукуямы.
Лечение амиотрофии Верднига-Гоффмана
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар. В тяжелых ситуациях (например, при выраженной гипоксемии вследствие слабости дыхательных мышц) пациентов переводят в отделение реанимации и интенсивной терапии и подключают к аппарату ИВЛ. На сегодняшний день не существует этиотропной терапии амиотрофии Верднига-Гоффмана. Все мероприятия носят симптоматический и паллиативный характер. Применяются следующие методы лечения:
- Обеспечение питания. Так как процесс глотания у многих пациентов затруднен, особое внимание уделяется вопросу кормления. Консистенция пищи должна быть полутвердой, положение ребенка – вертикальным. При необходимости устанавливается назогастральный зонд.
- Физиотерапия. Для улучшения метаболизма в мышечных тканях проводятся сеансы электрофореза, электростимуляция модулированным током, грязевые аппликации.
- ЛФК. С целью повышения мышечного тонуса необходимы регулярные физические упражнения – пассивные (выполняются специалистом) с постепенным переходом на активные (выполняются самим пациентом).
- Ортопедическое лечение. Для борьбы с костно-суставными деформациями, а также для их предупреждения используются ортопедические приспособления (корсеты, ортезы, иммобилизирующие шины), фиксирующие различные части тела.
- Респираторная поддержка. Важное место в лечении занимает устранение кислородной недостаточности. В зависимости от тяжести состояния больного назначаются ингаляции кислорода через лицевую маску/назальную канюлю или неинвазивная вентиляция легких через портативные аппараты ИВЛ.
Для достижения максимального эффекта лечение должно быть комплексным, проводиться непрерывно и подбираться индивидуально для конкретного пациента. Лекарственные препараты, применяющие для лечения амиотрофии Верднига-Гоффмана следующие:
- Метаболические средства. Для улучшения метаболических процессов в нервных клетках и мышечной ткани назначается коэнзим Q10, L-карнитин, ноотропы. Также для стимуляции регенерации нервной ткани используют высокие дозы витаминов группы В (В1, В6, В12).
- Вальпроаты. Противоэпилептические препараты из группы производных вальпроевой кислоты способны увеличивать образование белка выживания мотонейронов (SMN), что впоследствии может улучшать клиническое течение заболевания.
- ИПП и прокинетики. Ингибиторы протонной помпы (пантопразол) и ЛС, ускоряющие моторику желудочно-кишечного тракта (итоприд), помогают в борьбе с гастроэзофагеальным рефлюксом, который часто возникает у больных АВГ вследствие выраженного нарушения глотания.
- Муколитики и отхаркивающие средства. С целью борьбы с такими дыхательными проблемами как слабое откашливание, скопление в дыхательных путях густой мокроты, применяются препараты разжижающие мокроту (ацетилцистеин) и стимулирующие ее отхаркивание (терпингидрат).
При развитии грубых деформаций грудной клетки и позвоночника или крайне выраженных контрактур суставов показаны ортопедические операции. У лежачих больных, страдающих постоянно рецидивирующими пневмониями, выполняется трахеостомия. При гастроэзофагеальном рефлюксе, резистентном к медикаментозному лечению, прибегают к лапароскопической фундопликации Ниссена.
Ведутся постоянные исследовательские работы по поиску эффективного лекарства для терапии спинальной мышечной атрофии. Наиболее перспективным направлением считается генная терапия. В клинической практике уже используются антисмысловые олигонуклеотиды, исправляющие дефекты матричной РНК в гене SMN2 (Спинраза).
В конце 2019 года был зарегистрирован и одобрен к клиническому применению препарат Золгенсма, который содержит функционально полноценный ген SMN1. Доставка этого гена в нервные клетки производится с помощью аденоассоциированного вируса, проникающего через гематоэнцефалический барьер. Использование Золгенсмы приводит к значительному увеличению продукции белка SMN1 и улучшению состояния пациентов.
Прогноз
Врожденная амиотрофия Верднига-Гоффмана имеет крайне неблагоприятный прогноз. При ее манифестации в первые дни жизни ребенка, его гибель, как правило, происходит до 6-месячного возраста. При начале клиники после 3-х месяцев жизни, летальный исход наступает в среднем к возрасту 2 года, иногда — к 7-8 годам. Ранняя детская форма характеризуется более замедленным прогрессированием, дети погибают в возрасте 14-15 лет.
Профилактика
Первичная профилактика амиотрофии Верднига-Гоффмана заключается в пренатальной диагностике. При обнаружении в биоптатах ворсин хориона или амниотической жидкости мутации SMN1 показано прерывание беременности. Вторичная профилактика сводится к предупреждению осложнений – аспирационной пневмонии, контрактур суставов, инфекций нижних дыхательных путей.
Читайте также:
