
Синдром изменения костей черепа
Поражения костей свода черепа часто выявляются случайно при различных исследованиях головы. Хотя чаще всего они доброкачественные, очень важно выявлять и точно идентифицировать первичные и метастатические злокачественные поражения свода черепа. В этой статье рассматривается анатомия и развитие свода черепа, дифференциальная диагностика как единичных, так и множественных поражений свода черепа. Представлены примеры этих поражений, обсуждаются основные особенности визуализации и клинические проявления.
Цель обучения: перечислить частые одиночные и множественные поражения и псевдопоражения костей свода черепа и описать их типичные радиологические и клинические признаки.
Calvarial Lesions and Pseudolesions: Differential Diagnosis and Pictorial Review of Pathologic Entities Presenting with Focal Calvarial Abnormalities
A. Lerner, D.A. Lu, S.K. Allison, M.S. Shiroishi, M. Law, and E.A. White
Череп можно разделить на две области: основание черепа и свод. Большая часть свода формируется через интрамембранозную оссификацию, тогда как основание черепа через энхондральную оссификацию. Интрамембранозная оссификация происходит из мезенхимальных стволовых клеток соединительной ткани, а не из хряща. У новорожденных мембранозные кости свода черепа разделены швами. В местах пересечения швы расширяются, формируя роднички. Передний родничок находится в месте пересечения сагиттального, коронарного и метопического шва. Задний родничок расположен в месте пересечения сагиттального и лямбдовидного шва. Задний родничок обычно закрывается первым на третьем месяце жизни, а передний родничок может оставаться открытым в течение второго года.
Псевдопоражения свода черепа
Во время радиологического исследования литических поражений следует иметь в виду хирургические дефекты, такие как фрезевые отверстия или дефекты после трепанации черепа и нормальные варианты, известные как псевдопоражения. Сравнение с предыдущими исследованиями, анамнез и клинические данные часто помогают в неясных случаях.
Теменные отверстия
Теменные отверстия – парные округлые дефекты в задних парасагиттальных отделах теменных костей около макушки. Эти дефекты вовлекают как внутреннюю, так и наружную пластики и часто пропускают кровеносные сосуды (Рис. 1).
Сосуды не постоянно присутствуют, но здесь могут проходить эмиссарные вены, впадающие в верхний сагиттальный синус и артериальные ветви. Эти отверстия формируются в результате аномалии интрамембранозной оссификации в теменных костях, поэтому размеры их сильно различаются. Прилегающие мягкие ткани головы всегда нормальные. Иногда встречаются гигантские теменные отверстия, отражающие различную выраженность нарушения оссификации. Хотя эти отверстия считаются доброкачественным состоянием, но они могут сочетаться с внутричерепными венозными сосудистыми аномалиями, выявляемые на КТ и МРТ.
Двустороннее истончение теменных костей является другим состоянием, встречающимся у пожилых людей. Это истончение обычно вовлекает диплоический слой и наружную пластику свода черепа, приводя к зубчатому виду, не связано с сосудистыми структурами.
Венозные лакуны
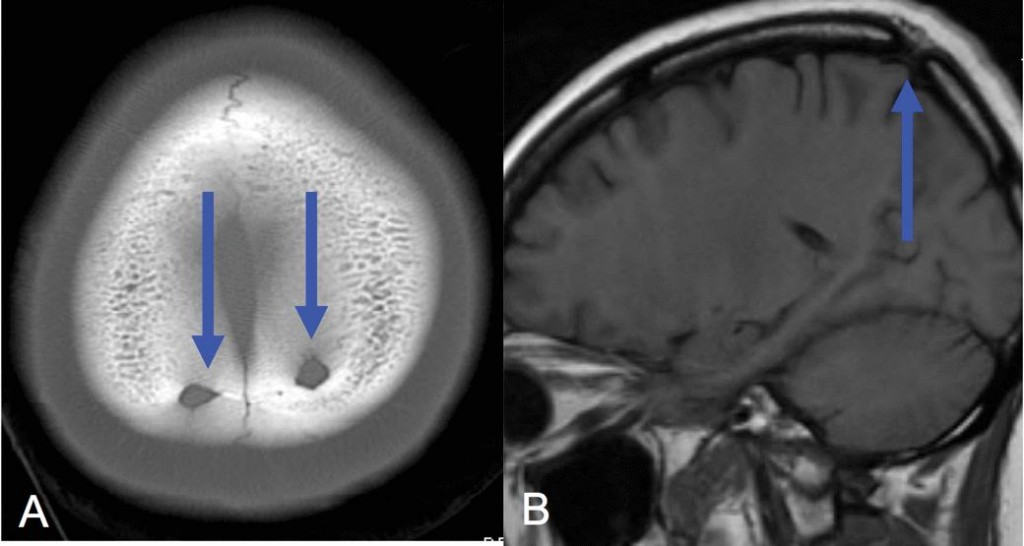
Венозные лакуны часто выявляются на КТ и рентгенограммах черепа в виде хорошо ограниченных овоидных или дольчатых фокусов просветления в костях свода черепа (Рис. 2).
Венозные лакуны – результат фокального расширения венозных каналов. На КТ часто видны расширенные дуральные венозные каналы без значительного вовлечения наружной пластинки костей свода черепа. МРТ и МР-венография могут демонстрировать расширенные сосуды в диплоическом слое.
Арахноидальные грануляции
Арахноидальные грануляции - это выпячивания арахноидальной оболочки и субарахноидального пространства в твердую мозговую оболочку, обычно в дуральные венозные синусы. Они обнаруживаются в поперечном синусе, в кавернозном синусе, в верхнем каменистом синусе и в прямом синусе. Пульсация ликвора может вызвать эрозию кости, выявляемую при визуализации.
На КТ арахноидальные грануляции изоденсивные ликвору, не накапливающие контраст округлые или овальные дефекты наполнения в синусах. На МРТ изоинтенсивные по отношению к ликвору. Они могут быть окружены костью или венозной пустотой потока и не накапливают контраст (Рис. 3). Дефект обычно вовлекает внутреннюю пластинку и диплоический слой и не затрагивает наружную пластинку.
Единичные поражения костей свода черепа
Дифференциация одиночного поражения от множественных может помочь в диагностике. Гемангиома, плазмоцитома, гемангиоперицитома, эпидермоидная киста, атретическое париетальное цефалоцеле могут быть единичными. Фиброзная дисплазия, остеома, внутрикостная менингиома и лимфома обычно бывают единичными, реже множественными. Поражения также разделяют на литические и склеротические.
Единичные литические доброкачественные и врожденные поражения
Эпидермоидная киста
Эпидермоидная киста - нечастое, доброкачественное медленно растущее образование. Она может быть врожденной или приобретенной, локализуется в любой части черепа, развивается от первой до седьмой декады жизни. Она обычно остается бессимптомной в течение многих лет, но изредка может подвергаться малигнизации в сквамозно-клеточную карциному. Хирургическое вмешательство показано для косметического эффекта, предупреждения неврологического дефицита и малигнизации. На КТ эпидермоидная киста обычно изоденсивна ликвору с хорошо отграниченным склеротическими краями (Рис. 4).
Кальцинаты встречаются в 10%-25% случаев. На МРТ киста изоинтенсивна или слегка гиперинтенсивна по отношению к серому веществу на Т1 и Т2 ВИ, гиперинтенсивна на FLAIR и DWI. Обычно значительного накопления контраста не отмечается. Дермоид предполагается при наличии жирового сигнала (гиперинтенсивный на Т1 и Т2).
Атретическое париетальное цефалоцеле
Атретическое париетальное цефалоцеле – это подапоневротическое образование, состоящее в основном из мягкой мозговой оболочки. Это абортивная форма цефалоцеле, распространяется через наружную и внутреннюю пластики черепа к твердой мозговой оболочке. Эта патология может сочетаться с другими внутричерепными аномалиями и плохим прогнозом с задержкой психического развития и ранней смертью.
Это поражение изначально кистозное, но может сглаживаться и сочетаться с алопецией в прилегающей коже. Существует также сочетание с персистирующей вертикальной веной фалькса, которая может иметь вид аномально расположенного эквивалента вертикального прямого синуса. Ликворный тракт, указывая на поражение, может распространяться через фенестрированный верхний сагиттальный синус (Рис. 5). На КТ видна подкожная киста или узел, изоденсивный ликвору. Узел может накапливать контраст за счет аномальных сосудов.
Гемангиома
Однако большие образования могут быть гипоинтенсивными на Т1. При кровоизлиянии в гемангиоме интенсивность сигнала может быть различной, зависящей от возраста кровоизлияния.
Единичные литические опухолевые поражения свода черепа
Плазмоцитома
Плазмоцитома – плазмоклеточная опухоль, которая может развиваться в мягких тканях или в структурах скелета. Самая частая локализация в позвонках (60%). Может также быть в ребрах, черепе, костях таза, бедре, ключице и лопатке. Пациенты с плазмоцитомой обычно на 10 лет моложе пациентов с множественной миеломой. На КТ определяется литическое поражение с зазубренными, плохо отграниченными несклерозированными контурами. Накопление контраста в них от слабого до умеренного. На Т1 ВИ гомогенный изоинтенсивный или гипоинтенсивный сигнал, на Т2 ВИ также изоинтенсивный или умеренно гиперинтенсивный сигнал в месте поражения (Рис. 7). Иногда может встречаться сосудистая пустота потока. Небольшие поражения могут быть в диплоическом слое, в больших очагах обычно определяется деструкция внутренней и наружной пластинки.
Гемангиоперицитома
Внутричерепная гемангиоперицитома – опухоль, исходящая из мозговых оболочек, растущая из перицистов, происходящих из клеток гладкой мускулатуры, окружающей капилляры. Гемангиоперицитома это гиперваскулярное образование из твердой мозговой оболочки, рентгенологически похожее на менингиому, но другое гистологически. Она высококлеточная, состоит из полигональных клеток с овальными ядрами и скудной цитоплазмой. Типичные спирали и псаммомные тельца, обнаруживаемые в менингиомах, отсутствуют. Часто выявляется сопутствующая фокальная деструкция черепа. Эти опухоли могут развиваться из примитивных мезенхимальных клеток по всему телу. Чаще всего в мягких тканях нижних конечностей, таза и забрюшинного пространства. Пятнадцать процентов возникает в области головы и шеи. Они составляют 0,5% от всех опухолей ЦНС и 2% от всех менингиальных опухолей. При визуализации выявляются дольчатые, накапливающие контраст экстрааксиальные опухоли, связанные с твердой мозговой оболочкой. Чаще всего локализуются супратенториально в затылочной области, обычно вовлекается фалькс, тенториум или дуральные синусы. Размеры могут быть разными, но чаще около 4 см. На КТ определяется экстрааксиальное образование повышенной плотности с перифокальным отеком и кистозным и некротическим компонентом пониженной плотности (Рис. 8).
Кроме деструкции костей свода может определяться гидроцефалия. Гемангиоперицитома может быть похожа на менингиому без кальцинатов и гиперостоза. На МРТ обычно определяется образование изоинтенсивное серому веществу на Т1 и Т2, но с выраженным неоднородным контрастированием, внутренней пустотой потока и очагами центрального некроза.
Лимфома
Лимфомы составляют до 5% от всех злокачественных первичных опухолей костей. Около 5% внутрикостных лимфом возникают в черепе. Важно отличать первичные от вторичных форм, которые имеют худший прогноз. Первичная лимфома относится к единичным опухолям без признаков отдаленных метастазов в течение 6 месяцев после выявления. КТ может выявить костную деструкцию и вовлечение мягких тканей. Лимфома может быть инфильтративной с деструкцией внутренней и наружной пластинок. На МРТ определяется низкий сигнал на Т1 с гомогенным контрастированием, на Т2 неоднородный сигнал от изоинтенсивного до гипоинтенсивного и снижение диффузии (Рис. 9).
Единичные склеротические поражения свода черепа
Фиброзная дисплазия
Фиброзная дисплазия – поражение кости с замещением нормальной костной ткани фиброзной тканью. Как правило, выявляется в детстве, обычно до 15 лет. Основание черепа - частая локализация краниофасциальной фиброзной дисплазии. Типичный КТ-признак это матрикс в виде матового стекла (56%) (Рис. 10). Однако может быть аморфное понижение плотности (23%) или кисты (21%). В этих участках может быть патологический трабекулярный паттерн, похожий на отпечатки пальцев. Усиление на КТ трудно оценить, за исключением участков пониженной плотности. На МРТ фиброзная дисплазия имеет низкий сигнал на Т1 и Т2 в оссифицированных и фиброзных участках. Но сигнал часто неоднородный в активную фазу. Пятнистый высокий сигнал на Т2 соответствует участкам пониженной плотности на КТ. На постконтрастных Т1 ВИ может быть накопление контраста.
Остеома
Остеома – доброкачественный костный вырост мембранозных костей, часто вовлекающий околоносовые синусы и кости свода черепа. Чаще всего встречается на шестой декаде жизни, соотношение мужчины/женщины 1:3. Множественные остеомы позволяют заподозрить синдром Гарднера, который характеризуется развитием множественных колоректальных полипов с возможной малигнизацией и внекишечных опухолей, включая остеомы. При визуализации остеома - хорошо отграниченное склеротическое образование с ровными контурами. На рентгенограммах и КТ обычно видно округлое склеротическое образование из наружной пластики костей черепа без вовлечения диплоического слоя (Рис. 11). На МРТ определяется хорошо отграниченная зона разрежения кости с низким сигналом на Т1 и Т2 ВИ без значимого накопления контраста. Другие доброкачественные мезенхимальные опухоли черепа, такие как хондрома и остеохондрома обычно вовлекают основание черепа.
Менингиома
Первичная внутрикостная менингиома редкая опухоль. Происхождение менингиом свода черепа неоднозначно. Опухоли могут происходить из эктопических менингоцитов или возможно из арахноидальных верхушечных клеток, запертых в черепных швах. Самый частый признак - растущее образование под кожей головы (89%), другие признаки: головная боль (7,6%), рвота и нистагм (1,5%).
На КТ определяются проникающие склеротические изменения в пораженной кости, в 90% с выраженным гомогенным контрастированием. Внекостный компонент поражения изоинтенсивен серому веществу на Т1 и изоинтенсивен или слабо гиперинтенсивен на Т2 с ярким контрастированием и иногда с участками низкого сигнала в кальцинатах (Рис. 12 и 13).
Типичные дуральные менингиомы часто вызывают гиперостоз в прилегающих костях черепа без прямой костной инвазии.
Множественные поражения свода черепа
Обычно это болезнь Педжета, гиперпаратироидизм, метастазы, множественная миелома, гистиоцитоз из клеток Лангенгарса. Они могут быть множественные или диффузные и с поражением других костей скелета. Редко они могут быть единичными поражениями костей черепа, но обычно на момент диагностики есть и другие костные поражения.
Болезнь Педжета
Болезнь Педжета чаще всего возникает у людей старше 40 лет. Обычно болезнь Педжета развивается в три стадии. Остеолизис возникает на ранней стадии в результате преобладания активности остеокластов в пораженной кости. Остеопороз circumscripta - большое литическое поражение на ранней стадии, вовлекающее внутреннюю и наружную пластики. (Рис. 14). Во вторую стадию развивается активность остеобластов, что приводит к восстановлению кости с участками склероза с типичным видом клочков ваты. В поздней стадии преобладает остеосклероз с обезображенными костными трабекулами и утолщением костей свода.
На КТ определяется диффузное гомогенное утолщение основания и свода черепа. Болезнь Педжета обычно не поражает кости носа, пазух и нижнюю челюсть.
На МРТ низкий сигнал на Т1 из-за замещения костного мозга фиброзной тканью, на T2 с высоким разрешением патологически высокий сигнал. Утолщенный свод черепа обычно неоднородно накапливает контраст (Рис. 15).
Гиперпаратироидизм
Метастазы
Метастазы свода черепа относятся к диффузным метастатическими поражениями скелета. Твердая мозговая оболочка является барьером для распространения опухолей из костей свода и эпидуральных метастазов. 18 КТ лучше выявляет эрозии основания черепа и внутренней пластинки, а МРТ более чувствительно для выявления распространения в полость черепа. Радионуклидные исследования костей можно использовать как скрининг для выявления костных метастазов. 18 КТ выявляет фокальные остеолитические и остеобластические поражения диплоического слоя с вовлечением внутренней и наружной пластинки (Рис. 17).
На МРТ метастазы обычно гипоинтенсивны на Т1 и гиперинтенсивны на Т2 с выраженным контрастированием (Рис. 18). Они могут быть единичными и множественными.
Множественная миелома
Гистиоцитоз из клеток Лангерганса
Гистиоцитоз из клеток Лангерганса редкое заболевание, с участием клональной пролиферации клеток Лангерганса, может проявляется множественными очагами в костях черепа и реже солитарным очагом. Другие частые локализации в костях: бедренная кость, нижняя челюсть, ребра, и позвонки. 20 Самый частый симптом увеличивающееся мягкое образование черепа. Но солитарные очаги могут быть бессимптомными и случайно обнаруживаться на рентгенограммах. 20 На рентгенограммах определяются круглые или овальные хорошо отграниченные очаги просветления со скошенными краями.
На КТ определяется мягкотканное образование с литической деструкцией, различной во внутренней и наружной пластинке, часто с мягкотканной плотностью в центре. На МРТ определяется от низкой до средней интенсивности сигнала на Т1, гиперинтенсивный сигнал на Т2 и значительное накопление контраста. На МРТ также может быть утолщение и контрастирование воронки гипофиза и гипоталамуса. Рис 20.
Диффузное утолщение костей свода черепа
Утолщение свода неспецифическое состояние, встречающееся как нормальный вариант, связанное с дискразиями крови, хронической шунтирующей хирургией, акромегалией и терапией фенитоином. На рентгенограммах и КТ можно увидеть диффузное утолщение костей свода черепа (Рис. 21). Корреляция с анамнезом и применение фенитоина может объяснить причину утолщения костей.
Побочный эффект фенитоина, приводящий к диффузному утолщению свода черепа широко освещается. Фенитоин стимулирует пролиферацию и дифференциацию остеобластов через регуляцию преобразования фактора роста-1 и костных морфогенетических белков. Если утолщение костей асимметричное или сочетается с литическими или склеротическими участками, следует думать о другой этиологии, включая болезнь Педжета, диффузные костные метастазы, фиброзную дисплазию и гиперпаратироидизм.
Выводы
Поражения костей свода черепа часто встречаются в клинической практике. Правильная диагностика может быть трудной. Знание нормальных вариантов и дифференциальная диагностика поражений свода черепа важна при первичной диагностике. Рентгенологические и клинические особенности этих поражений и соответствующая дальнейшая диагностика с помощью других модальностей может помочь установить вероятный диагноз.
Некоторые неврологические расстройства сопряжены с системными заболеваниями костей, с которыми в связи с этим должен быть знаком врач-невропатолог, поэтому ниже приводится краткая информация о такого рода костной патологии.
Для фиброзной остеодисплазии,или болезни Брайцева-Лихтенштайна,характерно нарушение костеобразующей функции мезенхимы, проявляющееся в одной или нескольких костях, что ведет к их деформации и образованию в них очагов разрежения, обычно отграниченных от здоровой ткани кости склеротической каймой. Объем пораженной кости при этом может быть увеличен. Чаще поражаются трубчатые кости, но характерные изменения могут отмечаться и в костях черепа. В таких случаях возможны облитерация придаточных полостей носа, деформация глазниц, сужение отверстий в основании мозгового черепа и в лицевом черепе, ведущее к нарушению функции проходящих через них нервов и сосудов. Заболевание, возможно, наследственное, проявляется с детских лет. Описал в 1927 г. отечественный хирург В.Р. Брайцев (1878-1964), несколько позже - американский патологоанатом L. Liechtenstein (1906-1977).
Мраморная болезнь (болезнь Альберс-Шенберга)- семейный генерализованный остеосклероз, протекающий с лейкемической реакцией крови у детей, с анемией и лейкопенией у взрослых, нередко с атрофией зрительных нервов и глухотой. Характерны деформация мозгового и лицевого черепа, заращение придаточных полостей носа плотной бесструктурной костной тканью. Ввиду постепенного сужения отверстий в черепе и межпозвонковых отверстий могут возникать полиморфные проявления поражения периферической нервной системы как на черепном, так и на позвоночном уровнях. В позвонках костные балки губчатого вещества утолщены и уплотнены. В трубчатых костях отмечается сужение, а затем и исчезновение костномозговых полостей, эпифизы булавовидно утолщены и поперечно исчерчены, имеется склонность к патологическим переломам. Наследуется по аутосомно-рецессивному типу и тогда, проявляясь в фенотипе в первые годы жизни, быстро приводит к смерти, или же - по аутосомно-доминантному типу, проявляясь в 20-40-летнем возрасте. Описал болезнь в 1907 г. H.E. Abers-Schonberg.
Синдром Олбрайтапредставляет собой множественную фиброзную дисплазию костей, сопровождающуюся болями и спонтанными переломами; при этом возможны повреждения верхней стенки глазницы. В таких случаях отмечается односторонний экзофтальм, на той же стороне - атрофия зрительного нерва, офтальмопарез. Обычны головная боль, расстройство слуха, судороги, олигофрения, гипертиреоз, зоны кожной гиперпигментации. Проявляется в детском возрасте. У девочек при этом возможно преждевременное половое созревание (менструации начинаются в 5-8 лет). Этиология неизвестна. Описали синдром в 1937 г. американский эндокринолог F. Albright (род. в 1900 г.) и соавт.
Энцефалоофтальмическая семейная дисплазия Краузе-Ризе- эктомезодермальная дисплазия, проявляющаяся сразу после рождения главным образом неврологическими и офтальмологическими симптомами. Характерны долихоцефалия, иногда гидроцефалия, затылочная или пояснично-крестцовая грыжа, мозжечковая атаксия, абсансы, олигофрения, раздражительность, а также птоз верхних век, страбизм, миопия, отслойка сетчатки, катаракта. Возможны расщепление верхней губы, твердого нёба, врожденные пороки сердца и другие дефекты развития. Наследуется по аутосомно-доминантному типу. Описали
эту форму патологии в 1946 г. австрийский врач A.C. Krause и в 1958 г. американский офтальмолог A.B. Reese.
Краниометафизарная дисплазия- диффузное разрастание костной ткани черепа и метафизов трубчатых костей. Характерны большая голова, гипертелоризм, седловидный нос, широко расставленные зубы. Сужение отверстий основания черепа может обусловить поражение черепных нервов и сосудистые расстройства. Ноги обычно непропорционально длинные, их суставные зоны утолщены. Течение заболевания медленно прогрессирующее. Наследуется по аутосомно-рецессивному типу. Описал этот патологический процесс в 1957 г. O. Lehman.
Синдром Дзержинского- семейная гиперпластическая периостальная дистрофия, проявляющаяся комбинацией пороков развития, при этом характерны различные варианты краниосиностоза, базилярная импрессия. Кости мозгового черепа и лица утолщены, уплотнены, нос резко выступающий, утолщены ключицы, грудина, иногда наблюдается воронкообразная грудь, пальцы короткие, их фаланги утолщены. Синдром, вероятно, наследственный. Описал заболевание в 1913 г. польский врач В.Э. Дзержинский.
Синдром Ван-Бюхема- наследственный генерализованный гиперостоз, проявляющийся после наступления половой зрелости умеренными признаками акромегалии. С 3-го десятилетия жизни появляются экзофтальм, ухудшение слуха, периферические парезы лицевых нервов. На рентгенограммах отмечаются проявления генерализованного гиперостоза, в крови - повышение уровня щелочных фосфатаз, нормальное содержание кальция и фосфора. Описал синдром в 1952 г. голландский терапевт F. van Buchem.
Гипопластическая хондродистрофияпредставляет собой врожденную болезнь, характеризующуюся нарушением энхондрального остеогенеза. Характерны большой мозговой череп с выступающим затылком, седловидный нос, прогнатизм, низкий рост (у взрослых до 130 см) в основном за счет укорочения конечностей (микромиелический нанизм), короткие кисти, выражен поясничный лордоз. Возможны корешковые боли, нижний парапарез, обструктивные апноэ во сне. При рождении длина тела 46-48 см, отмечается значительное отставание моторного развития, возможно умеренное отставание умственного развития. На рентгенограммах выявляются диспропорция мозгового и лицевого черепа, уплощение основания черепа, укорочение трубчатых костей, утолщения подвздошных костей, крылья которых развернуты, сужение позвоночного канала. Тип наследования аутосомно-доминантный, в 80% случаев заболевание обусловлено новыми мутациями.
Черепно-мозговые грыжи
Врожденным пороком развития являются черепно-мозговые грыжи, которые встречаются с частотой 1:4000-5000 новорожденных. Эта форма порока развития формируется на 4-м месяце внутриутробного развития. Она представляет собой грыжевое выпячивание в области костного дефекта, который может быть различным по своему размеру и форме. Локализуются грыжи обычно в местах соединения костей черепа: между лобными костями, у корня носа, около внутреннего угла глаза (передние грыжи), в области соединения теменных костей и затылочной кости (задние грыжи). Чаще других встречаются передние черепномозговые грыжи. По локализации наружного отверстия грыжевого канала они дифференцируются на носолобные, носорешетчатые и носоглазничные. Задние черепно-мозговые грыжи разделяются на верхние и нижние в зависимости от того, где расположен дефект в затылочной области: выше или ниже затылочного бугра. Кроме названных вариантов черепномозговых грыж, иногда выявляются так называемые базальные грыжи, при которых имеется дефект костей основания черепа на дне передней или средней черепных ямок, и грыжевой мешок выпячивается в полость носа или носоглотки. Редко встречаются черепномозговые грыжи в области сагиттального шва.
Основными формами черепно-мозговых грыж являются: 1) менингоцеле,при которой грыжевой мешок представлен кожей и измененными мягкой и паутинными оболочками, твердая мозговая оболочка обычно не принимает участия в образовании грыжевого выпячивания, а фиксируется к краям дефекта кости; содержимым грыжевого мешка при этом является ЦСЖ; 2) менингоэнцефалоцеле- грыжевой мешок составляют те же ткани, а содержимое его, кроме ЦСЖ, составляет и ткань мозга; 3) менингоэнцефалоцистоцеле- грыжевое выпячивание, в которое, кроме тех же тканей, вовлекается и часть расширенного желудочка мозга. Из перечисленных трех форм черепно-мозговых грыж чаще встречается менингоэнцефалоцеле, нередко именуемое как энцефалоцеле. При гистологическом изучении грыжевого мешка и его содержимого выявляются утолщение и уплотнение (фиброз) мягкой и паутинной оболочек, резкая атрофия и перерождение оказавшейся в грыжевом мешке мозговой ткани.
Поверхность грыжевого выпячивания может быть покрыта неизмененной кожей или истонченной, рубцово-измененной кожей, имеющей синеватую окраску. Иногда уже при рождении ребенка в центре грыжи имеется ликворный свищ. Нередко в первые годы жизни ребенка размеры грыжевого выпячивания значительно увеличиваются, при этом его кожные покровы истончаются и изъязвляются. Возможен и разрыв грыжевого мешка с массивной ликвореей, опасной для жизни. К тому же изъязвления на поверхности грыжевого мешка и ликворные свищи зачатую инфицируются, что может обусловить развитие гнойного менингоэнцефалита. Грыжевое выпячивание бывает на ножке (заужено в основании) или же имеет широкое основание. В последнем случае оно нередко пульсирует, а при натуживании ребенка - напрягается. При пальпации грыжевое выпячивание может быть различной плотности, эластичным, флюктуирующим.
Передние черепно-мозговые грыжи вызывают обезображивание лица, деформацию глазниц, носа, при этом нередко отмечаются уплощенная широкая переносица, неправильное расположение глазных яблок, нарушение бинокулярного зрения. При назоорбитальных грыжах, как правило, выявляются деформация и непроходимость слезно-носового канала, часто развиваются конъюнктивит, дакриоцистит.Базальные черепно-мозговые грыжи, располагающиеся в полости носа или носоглотки, по внешнему виду напоминают полипы. Если грыжевой мешок находится в одной половине носа, возникает искривление носовой пере- городки; при этом дыхание затруднено, речь невнятная с носовым оттенком.
Очень крупные менингоэнцефалоцеле (есть описание передней черепно-мозговой грыжи диаметром 40 см) обычно сопровождаются выраженной мозговой патологией, и новорожденные в таких случаях оказываются нежизнеспособными. Судьба остальных больных, как правило, зависит от размеров и содержимого грыжевого выпячивания, а также возможности оперативного лечения этого порока развития. Дети нередко испытывают головную боль, головокружение. Очаговая мозговая симптоматика может отсутствовать или быть умеренно выраженной, однако возможны и очаговые неврологические симптомы, в частности центральные парезы, гиперкинезы, расстройства координации движений и пр., признаки недостаточности функций черепных нервов (I, II, VI, VII, VIII, XII). Возможны эпилептические пароксизмы, отставание умственного развития.
Черепно-мозговые грыжи могут сочетаться с другими врожденными аномалиями: микроцефалией, краниостенозом, гидроцефалией, микрофтальмией, эпикантусом, врожденным птозом верхнего века, аномалией развития сетчатой оболочки глаза и зрительных нервов, колобомами (дефекты тканей глазного яблока), врожденным гидрофтальмом, краниоспинальными аномалиями, расщеплением дужек позвонков.
Лечение мозговых грыж.Показаниями к безотлагательной операции у но- ворожденного являются ликворея из грыжевого мешка или быстрое увеличение размеров грыжи с истончением ее покровов и опасностью разрыва. При отсутствии срочных показаний к операции ребенок должен находиться под наблюдением педиатров, невропатологов, нейрохирургов, которые обычно совместно решают вопрос о возможности оказания больному нейрохирургической помощи и определяют наиболее благоприятные сроки операции. Надо иметь в виду, что оперативное лечение черепно-мозговой грыжи может быть эффективным и нередко приводит к благоприятному результату.
Противопоказаниями к операции являются воспалительные процессы в оболочках и в головном мозге, выраженные неврологические и психические расстройства (имбецильность, идиотия), проявления гидроцефалии, тяжелые сопутствующие уродства.
Хирургическое лечение заключается в выделении и иссечении грыжевого мешка с сохранением при этом его содержимого. Важными этапами операции являются герметичное зашивание твердой мозговой оболочки и тщательная пластика костного дефекта.
При сочетании носоглазничной грыжи и гипертелоризма выполняется сложная реконструктивная операция, включающая пластику костного дефекта и сближение глазниц. Затылочные мозговые грыжи могут содержать венозные синусы твердой мозговой оболочки, что необходимо иметь в виду при хирургическом вмешательстве.
Ксантоматоз. Ксантоматоз развивается вследствие резкого нарушения липоидного и холестеринового обмена. Многие авторы квалифицируют это заболевание как хронически протекающую форму ретикуло-эндотелиоза, так как в ретикулярных и эндотелиальных клетках откладываются жировые и липоидные вещества, что в дальнейшем приводит к образованию различной величины опухолевых узлов, которые очагово или диффузно поражают различные органы.
Впервые типичный случай костного ксантоматоза описал Ханд в 1893 г. В дальнейшем некоторые авторы в различное время описали отдельные формы этого заболевания.
Костным ксантоматозом страдают большей частью дети в возрасте до 4 - 5 лет и очень редко взрослые.
При костной форме в основном нарушается холестериновый об-мен. При этом заболевании наблюдается характерная триада симптомов: 1) деструкция костей - черепа, таза, лопаток, позвонков и бедер (рис. 59), очень редко длинных трубчатых костей; 2) одно или двусторонний экзофтальм; 3) несахарный диабет. У таких больных часто отмечаются тяжелые гингивиты, расшатывание и выпадение зубов, нередко глухота, ксантомные кожные высыпи, слизистые оболочки и кожные покровы большей частью имеют желтушную окраску. Офтальмологически, кроме экзофтальма, нередко наблюдаются параличи глазных мышц центрального происхождения, жировая дегенерация роговицы и сетчатой оболочки. В некоторых случаях может отсутствовать какой-нибудь один симптом, но деструкция костей черепа является постоянным признаком заболевания.
Обычно ксантоматозные опухолевые массы располагаются в костях черепа, а ксантомные клетки через гаверсовы каналы проникают в кости свода черепа, постепенно разрушая их. Когда поражаются лобные кости, в процесс вовлекаются и стенки глазниц. Ксантоматозные узлы заполняют полость глазницы, вследствие чего начинается выпячивание глазных яблок - экзофтальм. В зависимости от распространения процесса могут страдать зрительные нервы. Когда процесс идет из черепа в глазницы, зрительные нервы страдают редко, в исключительных случаях наблюдается атрофия; при этом имеет место смещение глазного яблока в ту или другую сторону. Экзофтальм прямо вперед бывает в тех случаях, когда процесс захватывает твердую мозговую оболочку зрительного нерва в интраорбитальной его части.
Ксантоматозные узлы, проникая на основание черепа, могут вовлекать в процесс гипофиз, производить давление на близлежащие центры в области третьего желудочка, что нередко приводит к появлению несахарного диабета. Последний особенно беспокоит больного, вызывая непрерывную жажду и большой диурез.
При данном заболевании иногда наблюдается отложение липоидов в легких, печени и селезенке, в связи с чем необходимо, кроме детального исследования костного скелета, производить рентгенографию легких
Рентгенологически при костной форме ксантоматоза определяется типичная картина, заключающаяся в наличии большого количества проникающих через всю толщу дефектов в костях черепа, которые располагаются в основном в теменной, височной и лобных костях. Дефекты обычно имеют неправильную округлую форму и изъеденные края. Иногда дефекты, сливаясь между собой, достигают очень больших размеров, захватывая целые участки. Такие же дефекты можно встретить в стенках глазниц и в тазовых костях.
Под нашим наблюдением находилось 3 детей в возрасте 3 - 4 лет с типичной костной формой ксантоматоза.
Несмотря на характерную клинико-рентгенологическую картину костного ксантоматоза, иногда возникают трудности при дифференциальной диагностике. При этом следует исключить костный туберкулез, который также сопровождается дефектами в костях черепа. Костные дефекты при туберкулезе не так многочисленны и не бывают таких больших размеров, контуры их смазаны, иногда наблюдаются секвестры, реактивные явления, творожистый распад, характерные свищи в области глазниц, чего не отмечается при ксантоматозе.
С. А. Рейнберг считает необходимым проводить дифференциальную диагностику еще с метастазами невробластомы, которая также встречается в раннем детском возрасте. Но при этом в половине всех случаев клинически определяется первичная опухоль живота, расположенная ретроперитонеально. Миелома и опухоль Юинга также могут метастазировать в череп, но они характеризуются другой клинической картиной.
Болезнь Педжета. В группу системных заболеваний входят различные формы фиброзной остеодистрофии черепа. У нас нет возможности останавливаться на описании всех форм остеодистрофий, да в этом и нет необходимости, так как офтальмолог встречается только с теми формами из них, при которых в той или иной мере появляются нарушения со стороны органа зрения. К таким заболеваниям может быть отнесена болезнь Педжета.
Деформирующий остит, который был описан Педжетом в 1876 г., раньше относили к редким заболеваниям. В настоящее время вследствие широкого применения рентгенологического метода эту болезнь стали значительно чаще диагностировать. Ею поражаются преимущественно лица пожилого возраста. По данным некоторых авторов, чаще заболевают мужчины. В. С. Майкова-Строганова и Д. Г. Рохлин на основании своих материалов считают, что мужчины и женщины заболевают одинаково часто.
Болезнь Педжета протекает почти бессимптомно. Больные жалуются на общее недомогание, боли в костях и мышцах. В большинстве случаев заболевание обнаруживается случайно, при рентгенологическом исследовании.
В основе этой болезни, как и при других остеодистрофиях, лежит перестройка костной ткани, при которой происходит резорбция костного вещества и замещение его новообразованной соединительной тканью; это приводит к обезображивающей деформации целых отделов костного скелета. Голова таких больных значительно увеличивается в размерах, вследствие утолщения и искривления нижних конечностей и сплющивания позвонков все туловище укорачивается. В. С. Майкова-Строганова и Д. Г. Рохлин указывают, что раньше всего поражаются тазовые, седалищные кости, затем крестцовые и тела позвонков. Изменения в бедренных и большеберцовых костях, а также в костях черепа наступают значительно позднее. Рентгенологическая картина при этом настолько характерна и оригинальна, что врач, увидевший больного и его рентгенограммы один раз, запоминает их на всю жизнь.
Кроме указанных изменений, иногда отмечается сужение зрительных отверстий, в связи с чем наблюдается атрофия зрительных нервов. Утолщение костей может привести к уменьшению объема глазниц, вследствие чего появляется экзофтальм. При вовлечении в процесс верхней глазничной щели могут поражаться проходящие через нее черепномозговые нервы, что приводит к различным парезам и параличам. Из-за утолщения костей может иногда суживаться и слуховой проход, что нередко обусловливает глухоту.
Хлорома глазницы. Хлорома глазницы относится к группе системных заболеваний кроветворного аппарата. В основе этого заболевания лежит острый миелоз.
Хлорома характеризуется одиночными или первично множественными опухолевидными разрастаниями зеленоватого цвета, расположенными в основном под периостом плоских костей. При этом поражаются преимущественно кости черепа, стенки глазницы, височная кость, также могут поражаться и ребра. Такие опухолевидные разрастания нередко обнаруживаются в селезенке, печени и лимфатических узлах. А. И. Абрикосов относит хлорому к проявлениям лейкемии, но при микроскопическом исследовании он находил картину крупноклеточной лимфосаркомы. Н. Н. Петров отмечал при хлороме высокий лимфоцитоз.
Данное заболевание наблюдается очень редко. До 1935 г. в мировой литературе было описано всего 80 случаев (Р. А. Батарчуков). В дальнейшем в нашей отечественной литературе были приведены 2 случая Ц. А. Пальцевой и 4 случая А. И. Покровским. В Государственном научно-исследовательском институте глазных болезней имени Гельмгольца за 28 лет наблюдалось только 2 случая; один из них был описан М. М. Балтиным.
Поражаются хлоромой лица любого возраста, но в основном дети старше 4 лет. Болезнь протекает злокачественно и быстро приводит к смерти. Для таких больных характерна восковидная окраска лица. Нередко наблюдается деформация головы, парез лицевых нервов. Постоянными признаками данного заболевания являются одно или двусторонний экзофтальм, ограничение подвижности глазного яблока и быстрое падение зрения.
Офтальмоскопически отмечаются застойные соски, кровоизлияния в сетчатку. Глазное дно имеет желтовато-оранжевую окраску (лейкемическое дно). В. Н. Архангельский считает эти изменения следствием расстройства циркуляции внутриглазных жидкостей.
Рентгенологическая картина большей частью малохарактерна, так как костные изменения не успевают развиться из-за того, что больные очень рано погибают. Иногда обнаруживаются солитарные или множественные костные дефекты в костях черепа и стенках глазницы. С. А. Рейнберг наблюдал при хлороме в стенках глазницы поднадкостничные корковые узелки, просветления и периостальные разращения. В наших случаях, несмотря на наличие большого экзофтальма и множественных опухолевидных узлов в позвоночнике, ребрах и внутренних органах, рентгенологически было обнаружено только диффузное затемнение глазницы без деструктивных изменений ее костных стенок.
Мраморная болезнь. Впервые это редкое заболевание было описано в 1940 г. Альберс-Шейнбергом. По данным С. А. Рейнберга, во всей мировой литературе до 1934 г. насчитывалось всего 50 случаев. За последние 20 лет ряд отечественных авторов приводят описание единичных случаев этого заболевания. За 30 лет существования рентгенологического отделения Института имени Гельмгольца отмечено лишь 3 случая данного заболевания.
Мраморная болезнь относится к системным страданиям, поражающим весь костный скелет. Этиология его еще окончательно не выяснена. Большинство авторов придерживаются того мнения, что в основе его лежит глубокое нарушение кальциевого обмена, которое ведет к неправильному процессу окостенения. Считают, что данное заболевание является наследственной дисплазией костной системы, передающейся по рецессивному признаку. Другие авторы указывают, что в основе этого заболевания лежит перестройка костного мозга за счет уменьшения его объема и замещения новообразованной костной тканью. Это нередко обусловливает полное заращение мозгового канала, вследствие чего развивается анемия, приводящая в дальнейшем больного к смерти.
Чаще всего мраморная болезнь проявляется в детском возрасте. У таких детей отмечается задержка роста, кожа и видимые слизистые оболочки бледны. Вследствие патологического состояния костного скелета кости хрупкие и ломкие, что ведет к частым переломам.
Клинически установить диагноз мраморной болезни не представляется возможным Большую помощь в этом отношении, как и при целом ряде других системных заболеваний, оказывает рентгенологический метод исследования.

Рис. 61. Мраморная болезнь. Рентгенограмма черепа в боковой проекции.
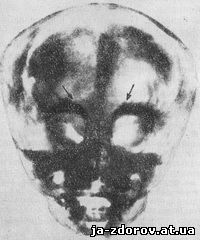
Рис. 62. Мраморная болезнь. Рентгенограмма черепа в передней проекции.
При рентгенологическом исследовании обнаруживается, что кости свода и основания черепа сильно уплотнены и проецируются в виде сплошных интенсивных теней, похожих на белый мрамор (рис. 61 и 62). Костные каналы, полости и отверстия сужены, иногда они зарастают вовсе. Нередко отмечается сужение зрительных отверстий, вызывающее понижение остроты зрения до полной слепоты. Придаточные пазухи носа при этом заболевании, особенно лобные, могут полностью зарастать плотной бесструктурной тканью. Такие же уплотнения наблюдаются и в других костях скелета. Только диафизы или метафизы длинных трубчатых костей сохраняют свое нормальное строение. На рентгенограммах видны светлые зоны, резко выступающие на фоне плотной кости, вследствие чего создается впечатление мраморности.
Рентгенологическая картина мраморной болезни настолько характерна, что дифференциальной диагностики не требуется.
Болезнь Крузона. Это заболевание, описанное впервые в 1912 г. Крузоном, своеобразно и встречается редко. Патогенез его до настоящего времени еще окончательно не выяснен.
Крузон, Бест, Фогт, Х. И. Бабенко и другие авторы считают, что в основе этого страдания лежит аномальное развитие костей черепа, мозговой и лицевой его части, обусловленное преждевременным зарастанием черепных швов, и относят его к семейной дистрофии. Почти все авторы высказываются за наследственный характер данного заболевания, но наряду с этим отмечают, что оно может появиться спорадически.
В связи с аномальным развитием костей черепа (как мозговых, так и лицевых) наблюдается характерная рентгенологическая картина.
Вследствие аномального развития костей черепа образуется различной величины экзофтальм с дивергенцией глазных яблок. Иногда экзофтальм настолько велик, что глазные яблоки почти выпадают из глазниц. При этом заболевании Фогт отмечал наличие симптома несмыкания челюстей и выпадения языка.
Офтальмоскопически выявляются застойные соски или атрофия зрительных нервов, наступающая вследствие застоя. Иногда может иметь место и первичная атрофия. Последняя возникает в результате сужения каналов зрительного нерва. Некоторые авторы отмечали при этом заболевании нистагм и катаракту.
Х. И. Бабенко наблюдала 2 больных с типичной лицевой формой болезни Крузона, у которых глазные симптомы отсутствовали. Такие же случаи описаны и другими авторами.
Читайте также:
