
Перонеальная мышечная атрофия болезнь шарко-мари
Перонеальная мышечная атрофия (болезнь Шарко-Мари-Тута) - это заболевание представляет собой наиболее распространенную форму генетически детерминированных невропатий; распространенность 3,8:100 000 населения. Тип наследования аутосомно-доминантный, экспрессивность составляет 83 %. Аномальный ген картирован в локусе 17р11.2. Продукт гена — протеин Р22 периферического миелина (РМР22). Встречающаяся значительно реже НМСН типа I с аутосомно-рецессивным типом наследования возникает в результате дефекта гена в локусе Xql3.1, что приводит к нарушению синтеза белка коннексин-32.
Клинические проявления болезни Шарко-Мари-Тута у детей. У большинства пациентов симптомы заболевания отсутствуют до позднего детского или раннего подросткового возраста. Однако у некоторых детей походка нарушается уже на втором году жизни. Большеберцовый и малоберцовый нервы поражаются наиболее рано, и их поражение наиболее выражено по сравнению с другими нервами. Дети с этим заболеванием кажутся неуклюжими, они часто спотыкаются и падают. Дебют симптомов может быть отсрочен до 6-го десятилетия жизни.
Мышцы передней поверхности голени атрофированы, и нога принимает характерную форму ноги аиста. Мышечная атрофия сопровождается прогрессирующей слабостью этих мышц, в связи с чем тыльное сгибание стопы затруднено, впоследствии формируется симптом свисающей стопы. Поражение билатеральное, но может определяться легкая асимметрия. Возможна деформация pes cavus (когтистая лапа) в связи с денервацией внутренних мышц стопы, что приводит к еще большему нарушению походки. Атрофия мышц предплечий и кистей обычно выражена не настолько сильно, как атрофия мышц нижних конечностей, при прогрессировании заболевания возможна контрактура лучезапястных суставов и пальцев рук, что приводит к формированию когтистой лапы. Слабость проксимальных мышц возникает на поздних стадиях заболевания и обычно умеренно выражена. Аксиальные мышцы йе поражаются.
Перонеальная мышечная атрофия медленно прогрессирует на протяжении всей жизни, однако в некоторых случаях двигательные нарушения быстро прогрессируют в течение нескольких лет. У большинства пациентов сохраняется способность к ходьбе и нормальная продолжительность жизни, хотя необходимо применение ортопедических приспособлений для стабилизации голеностопных суставов.
Среди чувствительных волокон поражаются главным образом крупные миелинизированные нервные волокна, передающие информацию о проприоцептивной и вибрационной чувствительности, возможно и повышение порога болевой и температурной чувствительности. Некоторые дети жалуются на ощущение покалывания или жжения в стопах, боль возникает редко. В связи с уменьшением мышечной массы нервы особенно уязвимы к травме или компрессии. Вегетативные проявления могут включать нарушение вазомоторного контроля с появлением красных пятен или бледности кожи стоп, при этом стопы необычно холодные на ощупь.
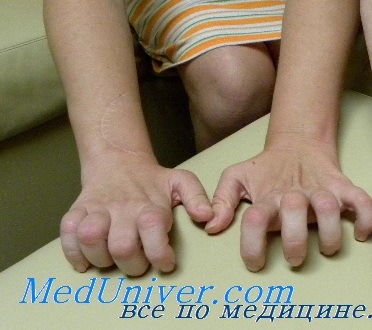
При пальпации нервы часто утолщены. Сухожильные рефлексы в дистальных отделах конечностей утрачены. Черепные нервы не поражаются. Функция тазовых органов не нарушена. При вегетативной невропатии отсутствует поражение сердца, ЖКТ и мочевого пузыря. Интеллект не снижен.
Синдром Давиденкова — вариант НМСН типа I с лопаточно-перонеальной атрофией и слабостью мышц.
Скорость проведения возбуждения по двигательным и чувствительным нервам значительно снижена, иногда достигает всего лишь 20 % нормального значения. При выявлении новых случаев заболевания при отсутствии семейного анамнеза необходимо обследование обоих родителей и исследование скорости проведения возбуждения по нервам.
Для подтверждения диагноза ЭМГ и мышечная биопсия обычно не требуются, однако эти методы обследования выявляют признаки многочисленных циклов денервации и реиннервации. Активность КФК в крови соответствует норме. В СМЖ возможно повышение уровня белка, цитоз отсутствует.
Разработан точный метод молекулярно-генетической диагностики по анализу крови.
Лечение болезни Шарко-Мари-Тута у детей. Основной проблемой является стабилизация голеностопных суставов. На ранних стадиях заболевания часто помогает ношение жесткой обуви высотой до середины голени, особенно когда пациент ходит по неровной поверхности, например по льду, снегу или по камням. В дальнейшем по мере развития слабости мышц, производящих тыльное сгибание стопы, возможно применение легких пластиковых шин для фиксации задней части голеностопного сустава и нижней части стопы. Они надеваются под носки и невидимы, что особенно удобно для пациентов. При развитии симптома свисающей стопы может потребоваться применение наружных фиксирующих ортопедических приспособлений в области нижней части ноги. В некоторых случаях рассматривается возможность хирургического укрепления голеностопного сустава.
Необходимо защищать ноги от травматического повреждения. В развернутой стадии заболевания можно предотвратить развитие компрессионной невропатии, подкладывая мягкие подушки под голени или между ними во время сна. Парестезия в виде жжения в стопах встречается редко, ее часто купируют приемом фенитоина и карбамазепина. Медикаментозное лечение, способное остановить или замедлить прогрессирование заболевания, не разработано.
Перонеальная мышечная атрофия - аксональный тип. По клинической картине это заболевание напоминает НМСН типа I, однако отличается меньшей скоростью прогрессирования и выраженностью двигательных нарушений. На ЭМГ денервация мышц. Биопсия икроножного нерва выявляет в большей степени аксональную дегенерацию, чем демиелинизацию и луковичные разрастания из шванновских клеток, характерные для НМСН типа I. Аномальный ген локализован на хромосоме 1 в локусе 1р35-р36. Это заболевание отличается от НМСН типа I, хотя оба заболевания наследуются по аутосомно-доминантному типу.
Болезнь Шарко-Мари-Тута болезнь Шарко Мари Тута, наследственная моторно-сенсорная невропатия I типа, наследственная нейропатия Шарко-Мари-Тута, ШМТ, невральная амиотрофия
Лечение болезни Шарко-Мари-Тута
Лечение назначается только после подтверждения диагноза врачом-специалистом. Показаны дозированная ЛФК и массаж, ортопедические мероприятия, витаминные препараты, средства нейротрофического действия, улучшающие микроциркуляцию, антихолинэстеразные препараты.
Имеются противопоказания. Необходима консультация специалиста.



- Аденозинтрифосфат натрия (средство, улучшающее метаболизм и энергообеспечение тканей). Режим дозирования: в/м, в первые 2-3 дня вводят 1 раз в день по 1 мл 1%-ного раствора, в последующие дни 2 раза в день или сразу 2 мл 1%-ного раствора 1 раз в день. На курс лечения — 30-40 инъекций.
- Пентоксифиллин (средство, улучшающее микроциркуляцию). Режим дозирования: внутрь, проглатывая целиком, во время или сразу после приема пищи, запивая достаточным количеством воды, в дозе 100 мг 3 раза в сутки с последующим медленным повышением дозы до 200 мг 2-3 раза в сутки.
- Мильгамма (комплекс витаминов группы В). Режим дозирования: терапию начинают с 2 мл внутримышечно 1 р/д на протяжении 5-10 дней. Поддерживающая терапия — 2 мл в/м два или три раза в неделю.
- Метандростенолон (анаболическое стероидное средство). Режим дозирования: внутрь, перед едой в дозе 0,005-0,01 г 1-2 раза в день. Курс лечения у взрослых длится 4-8 недель. Перерывы между курсами 4-8 недель.
- Церебролизин (ноотропное средство). Режим дозирования: применяют парентерально в виде в/м инъекций (до 5 мл) и в/в инъекций (до 10 мл). Препарат в дозе от 10 мл до 50 мл рекомендуется вводить только посредством медленных в/в инфузий после разведения стандартными растворами для инфузий. Продолжительность инфузий составляет от 15 до 60 мин. Вводят парентерально в дозе от 5 мл до 30 мл/сут. Рекомендуемый оптималь-ный курс лечения — ежедневные инъекции в течение 10-20 дней.
- Галантамин (антихолинэстеразное средство). Режим дозирования: внутрь, суточная доза для взрослых составляет 10-40 мг в 2-4 приема.
Формы болезни Шарко-Мари-Тута
Существует много форм болезни Шарко-Мари-Тута, включая CMT1, CMT2, CMT3, CMT4 и CMTX. CMT1, вызванный аномалиями в оболочке миелина, имеет три основных типа. CMT1A является аутосомно-доминантным заболеванием, которое возникает в результате дублирования гена на хромосоме 17, которая несет инструкции по производству периферического белка миелина-22 (PMP-22). Белок PMP-22 является критическим компонентом миелиновой оболочки. Сверхэкспрессия этого гена вызывает ненормальность структуры и функции миелиновой оболочки. Пациенты испытывают слабость и атрофию мышц нижних конечностей, начиная с подросткового возраста; позже они испытывают слабость рук и сенсорную потерю. Интересно, что другая невропатия, отличная от CMT1A, называемая наследственной невропатией с предрасположенностью к параличу давления (HNPP), вызвана удалением одного из генов PMP-22. В этом случае аномально низкие уровни гена PMP-22 приводят к эпизодической рецидивирующей демиелинизирующей нейропатии. CMT1B является аутосомно-доминантным заболеванием, вызванным мутациями в гене, который несет инструкции по изготовлению нуклеотида миелина (P0), который является еще одним критическим компонентом миелиновой оболочки. Большинство из этих мутаций являются точечными мутациями, что означает, что ошибка встречается только в одной букве генетического кода ДНК. На сегодняшний день ученые выявили более 120 различных точечных мутаций в гене P0. В результате аномалий в P0 CMT1B вызывает симптомы, сходные с симптомами, обнаруженными в CMT1A. Менее распространенные CMT1C, CMT1D и CMT1E, которые также имеют симптомы, сходные с симптомами, обнаруженными в CMT1A, вызваны мутациями в генах LITAF, EGR2 и NEFL соответственно.
Болезнь Шарко-Мари-Тута является результатом аномалий в аксоне периферической нервной клетки, а не в оболочке миелина. Он менее распространен, чем CMT1. CMT2A, наиболее распространенная аксоновская форма CMT, вызвана мутациями в Mitofusin 2, белке, связанном с митохондриальным слиянием. CMT2A также был связан с мутациями в гене, который кодирует белок 1B-бета-члена семейства кинезина, но в других случаях он не реплицируется. Кинезины — это белки, которые действуют как двигатели, чтобы помочь обеспечить транспортировку материалов вдоль клетки. Другие менее распространенные формы CMT2 были недавно идентифицированы и связаны с различными генами: CMT2B (связанный с RAB7), CMT2D (GARS). CMT2E (NEFL), CMT2H (HSP27) и CMT2l (HSP22).
CMT3 или Dejerine-Sottas — тяжелая демиелинизирующая невропатия, которая начинается в младенчестве. У младенцев происходит тяжелая атрофия мышц, слабость и сенсорные проблемы. Это редкое расстройство может быть вызвано определенной точечной мутацией в гене P0 или точечной мутацией в гене PMP-22.
CMT4 включает несколько различных подтипов аутосомно-рецессивного демиелинизирующего двигателя и сенсорных невропатий. Каждый подтип невропатии вызван другой генетической мутацией, может влиять на конкретную этническую популяцию и производит различные физиологические или клинические характеристики. Лица с CMT4 обычно развивают симптомы слабости ног в детстве, а в подростковом возрасте они не могут ходить. Несколько генов были идентифицированы как вызывающие CMT4, включая GDAP1 (CMT4A), MTMR13 (CMT4B1), MTMR2 (CMT4B2), SH3TC2 (CMT4C), NDG1 (CMT4D), EGR2 (CMT4E), PRX (CMT4F), FDG4 (CMT4H) и фиг.4 (CMT4J).
CMTX возникает точечной мутацией гена connexin-32 на Х-хромосоме. Коннексин-32-белок экспрессируется в клетках Schwann-клеток, которые обертывают нервные аксоны, составляя один сегмент миелиновой оболочки. Этот белок может участвовать в связи с клеткой Шванна с аксоном. Мужчины, которые наследуют один мутированный ген от своих матерей, проявляют умеренные или тяжелые симптомы заболевания, начиная с позднего детства или подросткового возраста (Y-хромосома, которую самцы наследуют от своих отцов, не имеет ген коннексин-32). Женщины, которые наследуют один мутированный ген от одного родителя и одного нормального гена от другого родителя, могут проявлять мягкие симптомы в подростковом возрасте или позже или вообще не могут развиваться симптомы заболевания.
Возможные осложнения ШМТ
Дыхание может быть затруднено, если болезнь поражает нервы, контролирующую диафрагму. Пациенту может понадобиться бронхолитические лекарственные средства или искусственная вентиляция легких. Избыточный вес или ожирение может затруднять дыхание.
Депрессия может быть результатом психического стресса, тревоги и разочарования жизни с любым прогрессирующим заболеванием. Когнитивная поведенческая терапия помогает пациентам лучше справляться с повседневной жизнью и, при необходимости, с депрессией.
Хотя ШМТ нельзя вылечить, некоторые меры помогут избежать дальнейших проблем. Они включают в себя хороший уход за ногами, так как существует повышенный риск травмы и инфекции, отказ от кофе, алкоголя и курения.
Понравилась новость? Читайте нас в Facebook
Приглашаем подписаться на наш канал в Яндекс Дзен
Как диагностировать ШМТ
Врач спросит о семейном анамнезе, и выявит признаки мышечной слабости — снижение мышечного тонуса, плоскостопие или высокий свод стоп (кавус).
Для исследования нервной проводимости проводится измерение силы и скорости электрических сигналов, которые проходят через нервы (Электромиография). Электроды помещаются на кожу, и вызывают слабые поражения электрическим током, которые стимулируют нервы. Задержанный или слабый ответ предполагает расстройство нервной системы, и, возможно, ШМТ.
При электромиографии (ЭМГ) тонкую иглу вводят в мышцы. Когда пациент расслабляет или сокращает мышцы, измеряется электрическая активность. Тестирование различных мышц покажет, какая из них страдает.
Генетическое тестирование проводится с помощью пробы крови, которая может показать, имеет ли пациент мутации гена.
Симптомы
Клиническая картина болезни имеет общие симптомы независимо от типа, но при этом может проявляться индивидуально. Даже в одной семье, когда заболевание провоцируется одним и тем же геном, у двух близких родственников оно далеко не всегда проявляется одинаково.
Общие симптомы болезни Шарко:
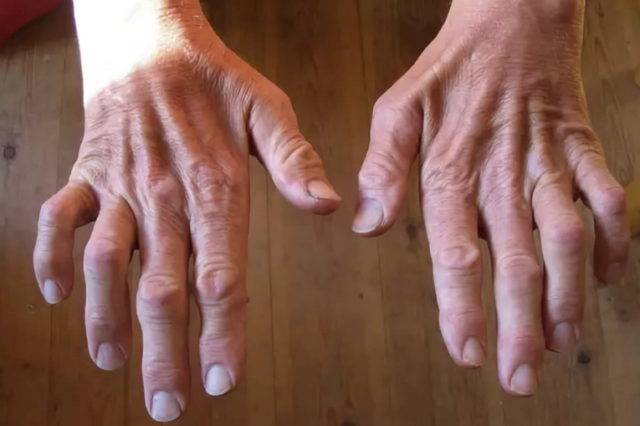
Дополнительные симптомы при 1-м типе болезни:
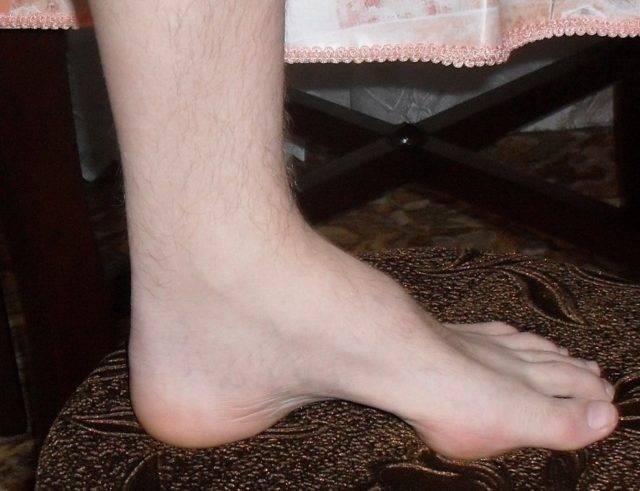
Особенности 2-го типа заболевания:
- изменения подъема стопы и формы пальцев встречаются реже, чем при 1-м типе;
- не наблюдаются утолщения нервных стволов;
- меньшая степень нарушения чувствительности;
- наблюдается синдром беспокойных ног перед сном;
- реже встречается ослабевание мышц в руках;
- возможно нарушение слуха (когда болезнь передается по женской линии);
- транзисторная энцефалопатия после физической активности на высоте: нарушения речи, шатание при ходьбе, затрудненное глотание, слабость проксимальных (ближних к туловищу) отделах конечностей.
Проявлением данной патологии обоих типов могут стать аутоиммунные реакции, когда вырабатываемые организмом специальные антитела разрушают миелиновые оболочки нервных волокон.
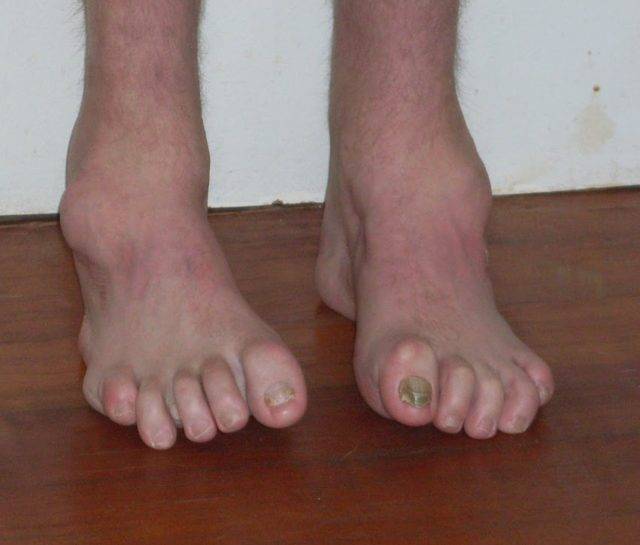
Симптомы болезни Шарко-Мари-Тута
Дебют заболевания отмечается в возрасте 10–20 лет. Первоначально появляются слабость в дистальных отделах ног, утомляемость в мышцах ног при длительном стоянии (постепенно нарастают на протяжении десятков лет). Позже может присоединяться боль в мышцах голени после долгой ходьбы (70%). При ходьбе приходится высоко поднимать ноги. Онемение в стопах отмечается в 80% случаев. Мышечная слабость в руках появляется спустя 10–15 лет после начала заболевания.
| Рис. 1 | Рис. 2 |
Диагностика
Диагностическая процедура включает ряд методов, среди которых:
- подробный личный и семейный анамнез;
- клиническая оценка мышечной силы, чувствительности;
- электрофизиологическое исследование скорости проводимости нервного волокна;
- неврологическое исследование.
Наиболее распространенные формы заболевания могут быть диагностированы путем анализа ДНК из крови пациента.
При диагностике необходимо тесное сотрудничество невролога, генетика, реабилитолога, хирурга-ортопеда и протезиста. В соответствии с выводами обследования, даются рекомендации относительно индивидуального плана реабилитации, при необходимости, назначается ортопедическая операция.
Значительная изменчивость клинических признаков болезни, наряду с отсутствием знаний о ней у многих врачей, часто приводит к неправильной диагностике.
Симптомы ШМТ у взрослых людей
- Слабость в мышцах ног и лодыжек;
- Искривление пальцев ног;
- Трудности подъема стопы из-за слабых мышц голеностопного сустава;
- Онемение в руках и ногах;
- Изменение формы голени, при этом нога становится очень тонкой ниже колена, в то время как бедра сохраняют нормальный объем мышц и форму (нога аиста);
- Со временем руки ослабевают и пациентам трудно выполнять повседневную работу;
- Появляются боли в мышцах и в суставах, человеку тяжело ходить. Нейропатическая боль возникает вследствие поврежденных нервов;
- В тяжелых случаях пациент может нуждаться в коляске, в то время как другие могут использовать специальная обувь или другие ортопедические устройства.
Факторы риска и причины ШMT
ШMT является наследственным заболеванием, так что люди, которые имеют близких родственников с заболеванием, имеют более высокий риск развития болезни.
Заболевание поражает периферические нервы. Периферический нервы состоит из двух основных частей: аксона — внутренняя часть нерва и миелиновой оболочки, которая является защитным слоем вокруг аксона. ШМТ может влиять на аксон и миелиновую оболочку.
При ШMT 1 мутируют гены, которые вызывают распад миелиновой оболочки. В конце концов, повреждается аксон, и мышцы пациента больше не получают четких сообщений от мозга. Это приводит к мышечной слабости и потере чувствительности или онемению.
При ШМТ 2 мутирующий ген влияет непосредственно на аксоны. Сигналы передаются не достаточно сильно, чтобы активизировать мышцы и органы чувств, так что пациенты имеют слабые мышцы, плохую чувствительность или онемение.
ШМТ 3 или болезнь Дежерин-Соттас, редкий тип заболевания. Повреждение миелиновой оболочки приводит к выраженной мышечной слабости и чувствительности. Симптомы могут быть заметны у детей.
ШМТ 4 является редким заболеванием, которое влияет на миелиновую оболочку. Симптомы обычно появляются в детстве, и пациенты часто нуждаются в инвалидном кресле.
ШМТ Х вызывается мутацией Х-хромосомы. Она чаще встречается у мужчин. Женщина с CMT X будет иметь очень слабые симптомы.
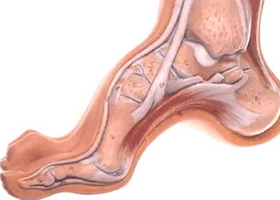
Общие сведения
Невральная амиотрофия Шарко Мари Тута объединяет группу наследственных прогрессирующих хронически протекающих полиневропатий:
- болезнь Рефсума;
- гипертрофическая невропатия Дежерина-Сотта;
- синдром Русси-Леви.
Частота встречаемости варьирует от 2 до 36 случаев на 100 тысяч населения. Невральная амиотрофия носит в основном семейный характер и у разных родственников клиническая симптоматика может сильно разниться. Параллельно регистрируются спорадические варианты течения болезни Шарко-Мари-Тута. Невральная амиотрофия чаще поражает лиц мужского пола.
Болезнь Шарко Мари Тута относится сразу к нескольким заболеваниям, которые получили название от имени Жана-Мартина Шарко:
Патогенез
Большинство форм болезни связано с поражением миелиновых оболочек в волокнах периферических нервов. Гораздо реже встречается патология аксонов (осевые цилиндры, которые проходят по центру нервного волокна). Дегенеративные изменения наблюдаются также в путях Голля (проводящая система глубокой чувствительности в спинном мозге), нейронах передних рогов, корешках спинного мозга (передние и задние), столбах Кларка (относятся к заднему спинномозжечковому пути).
На фоне дисфункции периферической нервной системы развивается мышечная атрофия, которая затрагивает отдельные группы миофибрилл. По мере прогрессирования патологии происходит смещение ядер сарколеммы, интерстициальное разрастание соединительной ткани, поражение миофибрилл. При нарастании гиалиновой дегенерации миофибрилл наблюдается их распад.
Классификация
Выделяют 2 типа невральной амиотрофии Шарко. Разграничение основывается на ряде особенностей, но в целом клиническая симптоматика у обоих типов схожа.
- I тип. Характерно выраженное снижение скорости проведения нервных импульсов. При биопсии нерва выявляется сегментарная демиелинизация нервных волокон и гипертрофическое разрастание непоражённых шванновских клеток.
- II тип. Скорость проведения импульсов практически не страдает, а при анализе биоптата выявляется дегенерация аксонов.
Также есть связь между атаксией Фридрейха и болезнью Шарко-Мари-Тута. У некоторых пациентов с ШМТ диагностируется клиническая симптоматика атаксии Фридрейха и наоборот (в течением времени). Также встречаются промежуточные формы этих заболеваний. В медицинской практике описаны случаи выявления атаксии Фридрейха у одних родственников и амиотрофии ШМТ – у других.
Причины
Достоверных данных о причинах развития невральной амиотрофии на сегодняшний день в практической неврологии нет. У 70-80% пациентов с болезнью Шарко отмечалось дублирование 1 участка 17 хромосомы. У патологии есть несколько форм, что обусловлено мутациями различных генов. Для заболевания характерен аутосомно-доминантный тип наследования с показателем пенетрантности на уровне 83%. Также были зарегистрированы случаи аутосомно-рецессивного типа наследования.
Симптомы болезни Шарко
В некоторых случаях заболевание начинается с нарушения чувствительности в стопах, и очень часто – с парестезий (чувство ползания мурашек по стопам). Характерным ранним признаком заболевания является отсутствие сначала ахилловых, а затем и сухожильных, коленных рефлексов. Часто пациенты предъявляют жалобы на болезненные, приступообразные сокращения в икроножных мышцах, которые усиливаются после длительной физической активности либо в ночное время.
Далее болезнь прогрессирует и поражает дистальные отделы рук – сначала кисти, а затем атрофия касается и мышц предплечий. Кисть становится похожей на лапу обезьяны из-за поражения тенара и гипотенара. Атрофические изменения никогда не регистрируются в мышцах плечевого пояса, туловища и шеи.
Очень часто отмечаются лёгкие фасцикулярные подёргивания мышц рук и ног. В некоторых случаях появляется компенсаторная гипертрофия в мышцах проксимальных отделов конечностей. Чувствительные нарушения проявляются тотальной гиперстезией. Болевая и температурная чувствительность страдают больше, чем глубокая. Редко появляется отёчность в поражённых конечностях и цианоз кожных покровов.
Клинические симптомы при болезни ШМТ прогрессируют очень медленно. Временной промежуток между поражением верхнего и нижнего пояса может достигать 10 лет. Длительное время пациенты остаются трудоспособными. Негативное влияние на скорость развития патологического процесса могут оказывать разные экзогенные факторы:
- черепно-мозговая травма;
- переохлаждение;
- перенесённые инфекции (краснуха, мононуклеоз, корь, ОРВИ, ангина);
- гиповитаминоз;
- травма позвоночника, спинного мозга.
Анализы и диагностика
Обследованием пациентов с подозрением на Болезнь Шарко-Мари-Тута занимаются невропатологи и ортопеды. При первичном осмотре уточняется возраст пациента, в котором впервые стала появляться характерная симптоматика. Обязателен сбор семейного анамнеза, с уточнением наличия у родственников схожих генетических заболеваний. При осмотре врач обращает внимание на деформацию кистей рук и стоп ног, на изменение походки.
Во время неврологического осмотра выявляется снижение тонуса в дистальных отделах верхних и нижних конечностей, снижение чувствительности кожных покровов и ослабление (вплоть до полного отсутствия) сухожильных рефлексов (коленные, ахилловы).
Основные методы исследования:
Дифференциальная диагностика проводится с заболеваниями:
- синдром Гийена-Барре;
- адренолейкодистрофия;
- спинальная мышечная атрофия Верднига-Гоффмана;
- болезнь Пелицеуса-Мерцбахера.
Лечение
Терапия пациентов с болезнью Шато проводится в стационарных условиях. Какого-либо специфического лечения нет, чтобы замедлить прогрессирование демиелинизации и аксональной дегенерации. Индивидуальная, грамотно подобранная терапия позволяет улучшить качество жизни пациента.

Невральная амиотрофия Шарко-Мари-Тута — это прогрессирующее хроническое наследственное заболевание с поражением периферической нервной системы, приводящем к мышечным атрофиям дистальных отделов ног, а затем и рук. Наряду с атрофиями наблюдается гипестезия и угасание сухожильных рефлексов, фасцикулярные подергивания мышц. К диагностическим мероприятиям относятся электромиография, электронейрография, генетическое консультирование и ДНК-диагностика, биопсия нервов и мышц. Лечение симптоматическое — курсы витаминотерапии, антихолинэстеразной, метаболической, антиоксидантной и микроциркуляторной терапии, ЛФК, массажа, физиопроцедур и водолечение.
МКБ-10

- Причины
- Патогенез
- Классификация
- Симптомы
- Осложнения
- Диагностика
- Лечение невральной амиотрофии Шарко-Мари-Тута
- Медикаментозная терапия
- Немедикаментозная терапия
- Хирургическое лечение
- Экспериментальное лечение
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Невральная амиотрофия Шарко-Мари-Тута (ШМТ) относится к группе прогрессирующих хронических наследственных полиневропатий, в которую входят синдром Русси-Леви, гипертрофическая невропатия Дежерина-Сотта, болезнь Рефсума и другие более редкие заболевания.
По различным данным, невральная амиотрофия Шарко-Мари-Тута встречается с частотой от 2 до 36 случаев на 100 тыс. населения. Зачастую болезнь носит семейный характер, причем у членов одной семьи клинические проявления могут иметь различную выраженность. Наряду с этим наблюдаются и спорадические варианты ШМТ. Лица мужского пола болеют чаще, чем женщины.
Причины
На сегодняшний день практическая неврология как наука не располагает достоверными сведениями о этиологии и патогенезе невральной амиотрофии. Проведенные исследования показали, что у 70-80% пациентов с ШМТ, прошедших генетическое обследование, отмечалось дублирование определенного участка 17-й хромосомы. Определено, что невральная амиотрофия Шарко-Мари-Тута имеет несколько форм, вероятно обусловленных мутациями различных генов. Например, исследователи выяснили, что при форме ШМТ, вызванной мутацией кодирующего митохондриальный белок гена MFN2, происходит образование сгустка митохондрий, нарушающего их продвижение по аксону.
Болезнь Шарко-Мари-Тута характеризуется аутосомно-доминантным наследованием с пенетрантностью на уровне 83%. Встречаются также случаи аутосомно-рецессивного наследования.
Патогенез
Установлено, что большинство форм ШМТ связаны с поражением миелиновой оболочки волокон периферических нервов, реже встречаются формы с патологией аксонов — осевых цилиндров проходящих в центре нервного волокна. Дегенеративные изменения затрагивают также передние и задние корешки спинного мозга, нейроны передних рогов, пути Голля (спинномозговые проводящие пути глубокой чувствительности) и столбы Кларка, относящиеся к заднему спинномозжечковому пути.
Вторично, в результате нарушения функции периферических нервов, развиваются мышечные атрофии, затрагивающие отдельные группы миофибрилл. Дальнейшее прогрессирование болезни характеризуется смещением ядер сарколеммы, гиалинизацией пораженных миофибрилл и интерстициальным разрастанием соединительной ткани. В последующем нарастающая гиалиновая дегенерация миофибрилл приводит к их распаду.
Классификация
В современной неврологической практике невральная амиотрофия Шарко-Мари-Тута подразделяется на 2 типа. Клинически они являются практически однородными, однако имеют ряд особенностей, позволяющих провести такое разграничение.
- Невральная амиотрофия I типа характеризуется существенным снижением скорости проведения нервного импульса. Биопсия нерва обнаруживает сегментарную демиелинизацию нервных волокон, гипертрофический рост непораженных шванновских клеток;
- При амиотрофии ШМТ II типа скорость проведения страдает незначительно, анализ биоптата показывает дегенерацию аксонов.
Отмечена связь болезни Шарко-Мари-Тута и атаксии Фридрейха. В отдельных случаях у пациентов с ШМТ со временем отмечаются типичные признаки болезни Фридрейха и наоборот — иногда по прошествии многих лет клиника атаксии Фридрейха сменяется симптоматикой невральной амиотрофии. Некоторыми авторами даны описания промежуточных форм этих заболеваний. Наблюдались случаи, когда у одних членов семьи диагностировалась атаксия Фридрейха, а у других — амиотрофия ШМТ.
Симптомы
В отдельных случаях невральная амиотрофия манифестирует расстройствами чувствительности в стопах, наиболее часто — парестезиями в виде ползания мурашек. Типичным ранним признаком ШМТ является отсутствие ахилловых, а позже и коленных сухожильных рефлексов. Основной симптом, на который пациенты чаще всего сами обращают внимание – приступообразные болезненные сокращения в икроножных мышцах (крампи), усиливающиеся в ночное время или после длительной физической нагрузки.
Развивающиеся первоначально атрофии затрагивают в первую очередь абдукторы и разгибатели стопы. Результатом является свисание стопы, невозможность ходьбы на пятках и своеобразная походка, напоминающая вышагивание лошади, — степпаж. Далее поражаются приводящие мышцы и сгибатели стопы. Тотальная атрофия мышц стопы приводит к ее деформации с высоким сводом, по типу стопы Фридрейха; формируются молоткообразные пальцы стопы. Постепенно атрофический процесс переходит на более проксимальные отделы ног — голени и нижние части бедер. В результате атрофии мышц голени возникает болтающаяся стопа. Из-за атрофии дистальных отделов ног при сохранности мышечной массы проксимальных отделов ноги приобретают форму перевернутых бутылок.
Зачастую при дальнейшем прогрессировании болезни Шарко-Мари-Тута атрофии появляются в мышцах дистальных отделов рук — вначале в кистях, а затем и в предплечьях. Из-за атрофии гипотенара и тенара кисть становиться похожей на обезьянью лапу. Атрофический процесс никогда не затрагивает мышцы шеи, туловища и плечевого пояса.
Часто невральная амиотрофия Шарко-Мари-Тута сопровождается легкими фасцикулярными подергиваниями мышц рук и ног. Возможна компенсаторная гипертрофия мышц проксимальных отделов конечностей. Сенсорные нарушения при невральной амиотрофии характеризуются тотальной гипестезией, однако поверхностная чувствительность (температурная и болевая) страдает значительно больше глубокой. В некоторых случаях наблюдается цианоз и отек кожи пораженных конечностей.
Для болезни Шарко-Мари-Тута типично медленное прогрессирование симптомов. Период между клинической манифестацией заболевания с поражения ног и до появления атрофий на руках может составлять до 10 лет. Несмотря на выраженные атрофии, пациенты длительное время сохраняют работоспособное состояние. Ускорить прогрессирование симптомов могут различные экзогенные факторы: перенесенная инфекция (корь, инфекционный мононуклеоз, краснуха, ангина, ОРВИ), переохлаждение, ЧМТ, позвоночно-спинномозговая травма, гиповитаминоз.
Осложнения
Невральная амиотрофия Шарко-Мари-Тута характеризуется ранней инвалидизацией. Вследствие прогрессирующей атрофии дистальных отделов конечностей и выраженных нарушений чувствительности больные постепенно теряют способность к самостоятельной ходьбе. Из-за грубых деформаций кистей рук пациенты не могут сами себя обслуживать. Контрактуры суставов нередко требуют хирургической коррекции.
На ранней стадии заболевания слабость в мышцах ног, гипестезия и гипорефлексия приводят к частым падениям, что повышает вероятность травм и переломов. Наиболее грозные неблагоприятные последствия происходят при сочетании болезни Шарко-Мари-Тута и атаксии Фридрейха. К ним можно отнести слепоту, кардиомиопатию, дыхательную недостаточность.
Диагностика
Курацией пациентов занимаются врачи-неврологи и ортопеды. При опросе больного уточняется возраст, в котором начали появляться симптомы (для болезни ШМТ типична манифестация в 15-25 лет). Важное значение имеет семейный анамнез (наличие близкого родственника с этой патологией). Во время общего осмотра обращается внимание на изменение походки, деформацию стоп и кистей.
При неврологическом осмотре отмечается уменьшение тонуса дистальных отделов верхних и нижних конечностей, ослабление или полное отсутствие сухожильных рефлексов (ахилловых, коленных), снижение кожной чувствительности. Для уточнения диагноза проводятся следующие методы исследования:
- ЭНМГ. При электронейромиографии отмечаются признаки аксональной и демиелинизирующей нейропатии – замедление скорости проведения импульса по двигательным нервам, падение амплитуды М-ответов.
- Компьютерная паллестезиометрия. Данная диагностическая процедура позволяет объективно оценить снижение вибрационной чувствительности – наиболее ранний признаки болезни ШМТ.
- Гистология. При гистологическом исследовании биоптата большеберцового нерва обнаруживаются уменьшение количества миелиновых волокон, разрастание соединительнотканных волокон, атрофию миелина.
- ДНК-анализ. Подтверждающий метод исследования, верифицирующий диагноз. Выявляются дупликации гена белка периферического миелина (PMP22) на 17-й хромосоме.
Дифференциальный диагноз невральной амиотрофии Шарко-Мари-Тута необходимо проводить с наследственными нейромышечными заболеваниями (спинальная мышечная атрофия Верднига-Гоффмана, адренолейкодистрофия, болезнь Пелицеуса-Мерцбахера) и приобретенными хроническими полинейропатиями (синдром Гийена-Барре).
Лечение невральной амиотрофии Шарко-Мари-Тута
Для прохождения лечения все больные подлежат обязательной госпитализации в стационар. В настоящее время не существует специфической терапии, способной замедлить прогрессирование аксональной дегенерации и демиелинизации. Однако своевременно начатая грамотная и индивидуально подобранная терапия способна значительно улучшить качество жизни пациентов. Из лекарственных препаратов для симптоматического лечения невральной амиотрофии ШМТ применяются:
- Витамины. Для улучшения микроциркуляции и восстановления нервных волокон назначаются инъекции витаминов группы В (В1, В3, В12). К витамину В6 стоит относиться с осторожностью, так как превышение его дозы оказывает нейротоксический эффект. По данным некоторых исследователей, аскорбиновая кислота способна подавлять образование периферического белка миелина (PMP22).
- Миорелаксанты. С целью устранения болезненных мышечных сокращений пациентам рекомендуется прием медикаментов, расслабляющих скелетную мускулатуру – баклофен, толперизон.
- Кальций и витамин Д. Так как примерно 40% больных имеют остеопороз, для уменьшения риска переломов им показаны препараты кальция и витамина Д (холекальциферол).
- Антихолинэстеразные средства. При болезни ШМТ 2 типа для улучшения нервно-мышечной проводимости целесообразно назначение прозерина, галантамина.
Основное внимание уделяется немедикаментозному лечению невральной амиотрофии Шарко-Мари-Тута. Для достижения максимального терапевтического эффекта применяется комплекс следующих мероприятий:
- Электростимуляция. Для усиления нейротрофики, активации метаболизма в паретичных мышцах и проводимости периферических нервов используется направленная подача электрических импульсов.
- ЛФК. С целью повышения мышечного тонуса рекомендуются регулярные занятия лечебной физкультурой. Наиболее эффективно совмещение активных (выполняются самим пациентом) и пассивных (выполняются специалистом) упражнений.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах (в первую очередь нижних конечностей) выполняются различные виды массажа – ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Бальнеотерапия. Грязевые ванны и грязевые аппликации способствуют коррекции нарушений вегетативной нервной системы и замедлению формирования контрактур.
- Ортопедическое лечение. Чтобы предупредить развитие грубых деформаций больным назначается ношение ортопедической обуви. При нестабильности суставов из-за мышечной слабости, для фиксации стоп в заданном положении используются специальные приспособления (ортезы, подтяжки).
Комплексное проведение данных мероприятий позволяет увеличить мышечную силу, исправить нарушения равновесия и походки. Благодаря этому удается повысить бытовую, социальную адаптацию, работоспособность пациентов.
При выраженных атрофических явлениях и деформации стопы, значительно затрудняющих самостоятельную ходьбу, когда консервативные методы оказываются безуспешными, показаны ортопедические оперативные вмешательства – метатарзальная остеотомия, остеотомия пяточной кости. В некоторых случаях для восстановления опорной функции стопы может понадобиться проведение артродеза.
Продолжаются поиски эффективного лекарства для борьбы с невральной амиотрофией Шарко-Мари-Тута. В клинических испытаниях, где пациенты принимали препарат PXT3003 (комбинация малых доз баклофена, налтрексона и сорбитола), были отмечены положительные результаты в виде увеличения мышечной силы, возобновления чувствительности и сухожильных рефлексов.
Рассматривается возможность использования в качестве лечения ингибиторов HDAC6 – ферментов, стимулирующих регенерацию белков цитоскелета нервных клеток. Эксперименты на лабораторных животных показали, что данные вещества способны значительно замедлить прогрессирование демиелинизации и аксональной дегенерации.
Прогноз и профилактика
Невральная амиотрофия Шарко-Мари-Тута – тяжелое инвалидизирующее заболевание. Большинство пациентов утрачивают способность ходить через 15-20 лет после начала появления симптомов. Однако в виду того, что преимущественно поражаются дистальные отделы конечностей, продолжительность жизни больных практически не отличается от таковой в общей популяции.
Летальные исходы в молодом и среднем возрасте наблюдаются при сочетании с атаксией Фридрейха, когда в патологический процесс вовлекается дыхательная мускулатура и миокард. Специфических методов первичной профилактики не существует. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
Читайте также:
