
Наследственные заболевания соединительной ткани клинические рекомендации
В статье изложены современные представления о терминологии и номенклатуре наследственных нарушений (дисплазий) соединительной ткани, приведены диагностические критерии отдельных клинических вариантов этой патологии.
Для преодоления существующих противоречий в дефинициях и критериях диагноза отдельных клинических вариантов ННСТ комитет экспертов Всероссийского научного общества кардиологов (ВНОК) разработал первые национальные рекомендации, принятые на Российском национальном конгрессе кардиологов в 2009 г. и пересмотренные в 2012 г. [2]. Эти усилия позволили существенно сблизить подходы к диагностике ННСТ в нашей стране с международной практикой.
Синдром Марфана — аутосомно-доминантная патология, в основе которой лежит мутации гена фибриллина-1 (FBN1). Фибриллин составляет основу эластических волокон; его особенно много в межклеточном матриксе сосудистой стенки, сердца, хрящей, хрусталика, роговицы и цинновой связки. Мутации гена FBN1 приводят к неполноценности фибриллина и нарушению структуры и функции перечисленных органов и тканей.
Диагностика синдрома Марфана основана на Гентских критериях (1996, 2010 гг.). В последней версии Гентских критериев [3] было упразднено деление на большие и малые признаки, ряд малых признаков исключен. Одновременно было выделено два наиболее специфичных признака — дилатация и/или расслоение аорты и эктопия хрусталика и предложена балльная оценка остальных признаков для расчета степени системного вовлечения соединительной ткани (СВСТ) — табл. 3. В отсутствие семейного анамнеза диагноз синдрома Марфана может быть установлен при наличии расширения корня аорты и эктопии хрусталика либо при сочетании расширения аорты с мутацией гена FBN1 или с совокупностью признаков СВСТ на 7 и более баллов. При отягощенном семейном анамнезе диагноз правомерен, если выявляется один из специфичных признаков или если СВСТ составляет 7 и более баллов.
Синдром Элерса–Данло — гетерогенная группа коллагенопатий с различными типами наследования и общими клиническими проявлениями в виде гипермобильности суставов и повышенной эластичности кожи. Диагностика синдрома Элерса–Данло основана на Вильфраншских критериях [4]. Вместо ранее признаваемых десяти типов болезни в настоящее время выделены шесть: классический, гипермобильный, сосудистый, кифосколиотический, артрохалазия, дерматоспараксис; для каждого из них определены большие и малые диагностические критерии. Для клинической диагностики необходимо наличие хотя бы одного большого критерия (табл. 4).
MASS-фенотип (или марфаноподобный синдром) — акроним, обозначающий пролапс митрального клапана (Mitral valve prolapse), расширение аорты (Aotic dilatation), изменения кожи (Skin) и костей скелета (Skeleton). MASS-фенотип можно диагностировать при пограничном расширении корня аорты, наличии хотя бы одного скелетного проявления и признаков СВСТ на 5 и более баллов. Как можно заметить, при отсутствии данных молекулярно-генетической диагностики MASS-фенотип трудно (если вообще возможно) отличить от синдрома Марфана с неполным набором признаков.
Пролапс митрального клапана диагностируется при систолическом смещении одной или обеих створок митрального клапана за линию клапанного кольца в парастернальной продольной позиции более чем на 2 мм. Морфологическим субстратом первичного пролапса митрального клапана как одного из вариантов ННСТ выступает миксоматоз створок, отражающий дезорганизацию коллагеновых фибрилл и накопление в них кислых гликозаминогликанов.
Первичный пролапс митрального клапана следует отличать от митрального пролапса как принадлежности моногенных ННСТ или MASS-фенотипа. Дифференциальными критериями (к сожалению, не абсолютными) являются диаметр аорты и количество признаков СВСТ.
В основе синдрома гипермобильности суставов лежат мутации генов, кодирующих коллаген, эластин, фибриллин и тенасцин Х, приводящие к слабости суставных связок. Синдром характеризуется избыточным диапазоном движений в суставах, сопровождающимся клинической симптоматикой (привычные вывихи, артралгии). При диагностике гипермобильности суставов используется девятибалльная шкала P. Beighton [5], предусматривающая оценку способности выполнения следующих пяти движений: пассивного сгибания V пястно-фалангового сустава более чем на 90°, пассивного приведения I пальца к предплечью, пассивного переразгибания коленных и локтевых суставов более 10°, свободного касания ладонями пола при прямых ногах. Первые четыре движения — парные (присваивается по баллу за возможность выполнить движение на каждой из сторон), последнее — непарное (максимально возможный суставной счет — 9 баллов). Гипермобильность суставов, составляющая не менее 4 баллов, и артралгии не менее чем в четырех суставах продолжительностью от трех месяцев и являются большими диагностическими критериями данной патологии.
Поскольку слабость связочного аппарата является универсальным признаком соединительнотканной недостаточности, синдром гипермобильности суставов исключается при наличии синдромов Марфана, Элерса–Данло и ряда других близких им по клиническим проявлениям ННСТ.
Неклассифицируемые ННСТ, не подходящие под согласованные критерии диагностики, встречаются в повседневной практике гораздо чаще. Многообразие их клинических вариантов систематизировано в следующие варианты: МASS-подобный фенотип, марфаноидная внешность, элерсоподобный фенотип, доброкачественная гипермобильность суставов, неклассифицируемый фенотип. Первые два из них фенотипически напоминают синдром Марфана, два следующие — синдром Элерса–Данло, не отвечая полностью критериям диагноза указанных состояний. В основу диагностики неклассифицируемых ННСТ положены те же принципы (совокупность внешних и висцеральных фенотипических проявлений), что используются при выявлении ННСТ, имеющих согласованные рекомендации, однако диагностический порог при этом менее высокий.
MASS-подобный (марфаноподобный) фенотип характеризуется пограничным значением размера корня аорты в сочетании с миопией и/или пролапсом митрального клапана и наличием признаков СВСТ менее 5 баллов (в отличие от MASS-фенотипа, при котором — 5 баллов и более).
Марфаноидная внешность характеризуется только признаками вовлечения костной системы (обычно у астеников) при отсутствии висцеральных изменений. При этом допускаются менее строгие скелетные изменения, чем те, что необходимы для констатации синдрома Марфана, однако наличие долихостеномелии и арахнодактилии признается обязательным.
Главное условие отнесения пациента к элерсоподобному фенотипу — наличие не менее двух признаков вовлечения кожи, исключая большие критерии синдрома Элерса–Данло.
Доброкачественная гипермобильность суставов констатируется на основе выявления избыточного диапазона движений в суставах, но без клинической симптоматики.
Литература
- Земцовский Э. В. Недифференцированные дисплазии соединительной ткани. Попытка нового осмысления концепции // Вестник медицины Северного Кавказа. 2008; 2: 8–14.
- Наследственные нарушения соединительной ткани в кардиологии. Диагностика и лечение. Российские рекомендации (I пересмотр) // Российский кардиологический журнал. 2013; 1 (Прил. 1): 1–32.
- Loeys B. L., Dietz H. C., Braverman A. C. et al. The Revised Ghent Nosology for the Marfan Syndrome // J. Med. Genetics. 2010; 4: 476–485.
- Beighton P., De Paepe A., Steinmann B. et al. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997 // Am. J. Med. Genetics. 1998; 1: 31–37.
- Grahame R., Bird H. A., Child A. The revised (Brighton, 1998) criteria for the diagnosis of benign joint hypermobility syndrome // J. Rheumatology. 2000; 7: 1777–1779.
А. В. Клеменов 1 , доктор медицинских наук
А. С. Суслов
ГБУЗ НО ГКБ № 30, Нижний Новгород
Abstract. The article is devoted to modern concepts of terminology and nomenclature of hereditary disorders of connective tissues. The authors adduce diagnostic criteria of particular clinical variants of this pathology.

- Причины возникновения
- Классификация заболевания
- Клинические проявления
- Диагностические мероприятия
- Лечение заболевания
- Изменение рациона питания
- Хирургические вмешательства
Причины возникновения

Дисплазия соединительной ткани у детей: проявления и подходы к лечению
Выделить одну причину развития дисплазии соединительной ткани у детей невозможно. Существуют различные факторы, способные привести к возникновению патологии. В зависимости от связи с наследственностью выделяют две формы дисплазии:
- недифференцированная, характеризующаяся выраженными клиническими проявлениями, но не имеющая наследственного характера;
- дифференцированная — связанная с генетическими изменениями.
Все факторы риска развития болезни делят на врожденные и приобретенные. К врожденным относят генетические мутации в генах, кодирующих белки соединительной ткани, например, коллагена или фибриллина. Приобретенные факторы представлены следующими состояниями: вредные привычки у матери во время беременности, нерациональное питание, негативные экологические условия и др.
Классификация заболевания
Для оценки прогноза и характера течения патологии врачи определяют тип дисплазии — дифференцированный или недифференцированный. Первая форма заболевания имеет установленную наследственную причину, например, синдром Марфана, Альпорта и др. Для диагностики дифференцированного варианта проводят генетические исследования у ребенка и родителей.
Недифференцированная дисплазия встречается чаще. Однозначно определить причину при этом не удается. Системные нарушения в структуре соединительной ткани приводят к различным клиническим проявлениям с поражением различных систем и органов. Для постановки подобного диагноза врач должен исключить наследственные синдромы.
Клинические проявления
Недифференцированная форма болезни не связана с наследственностью
Симптомы дисплазии в детском возрасте принято разделять на две группы: висцеральные и фенотипические. Они выявляются во время обследования у лечащего врача. Фенотипические признаки видны при внешнем осмотре. К ним относят:
- удлинение пальцев, стоп и кистей;
- различные варианты деформации грудной клетки;
- повышенная подвижность суставов. Например, ребенок может переразгибать локти или колени;
- появление варикозной болезни в подростковом возрасте;
- ранняя близорукость;
- ассиметричное лицо с нарушениями прикуса;
- искривления позвоночника различной степени тяжести;
- Х- и О-образная деформация голеней;
- множественные пигментные пятна и сосудистые сеточки на коже.
Указанные симптомы выявляются у большинства больных с дисплазией соединительной ткани. Они имеют разную степень выраженности.
В диагностике заболевания участвует не только педиатр, но и другие специалисты: кардиолог, пульмонолог, невролог и пр. Это зависит от клинической картины болезни.
Висцеральные проявления характеризуются изменениями работы внутренних органов и нервной системы на фоне дисплазии. Эти признаки не имеют внешних проявлений, поэтому специалисту необходимо тщательно собирать жалобы ребенка и его родителей. Основные висцеральные симптомы:
- плохой сон и повышенная сонливость в течение дня;
- частые головные боли, не имеющие конкретной локализации;
- гипервозбудимость;
- тревожность;
- нарушения работы органов желудочно-кишечного тракта в виде метеоризма, запоров и др.;
- изменение уровня артериального давления;
- признаки поражения мочевыделительной системы: ночное недержание мочи и т.п.
При выявлении указанных симптомов родителям необходимо сразу обратиться за медицинской помощью. Специалист подберет необходимое обследование и сможет поставить точный диагноз.
Диагностические мероприятия
Дисплазия соединительной ткани у детей выявляется при комплексном обследовании. Оно начинается с изучения имеющихся жалоб у ребенка и его родителей. После этого проводится внешний осмотр с оценкой степени подвижности суставов по шкале Бейтона. Врач проводит измерение длины стоп, рук и ног, а также замеры охвата головы и груди.
Проводятся дополнительные лабораторные и инструментальные методы. Для выявления заболеваний сердечно-сосудистой системы показано выполнение электрокардиографии, ЭхоКГ и УЗИ магистральных сосудов. Для оценки состояния тазобедренных суставов и других частей опорно-двигательного аппарата проводят рентгенографию или компьютерную томографию. При симптомах поражения внутренних органов назначают их дополнительное обследование.
Лечение заболевания
Терапия, позволяющая полностью избавиться от дисплазии соединительной ткани, не разработана. В основе патологии лежит врожденный или приобретенный генетический дефект, который невозможно исправить. Поэтому лечение направлено на предупреждение развития осложнений болезни и повышение качества жизни пациента.
В качестве лечебных мероприятий используют лекарственные препараты, физкультуру и изменения рациона питания. Терапия подбирается и проводится только под наблюдением лечащего врача.

Cимптомы заболевания различны в зависимости от формы патологии
В лечении применяют различные группы медикаментозных препаратов, улучшающих состояние внутренних органов и нервной системы. Основные используемые классы лекарственных средств:
- витаминные комплексы с добавлением магния. Обеспечивают нормализацию обмена веществ в организме и улучшают работу скелетной мускулатуры и сердца;
- ноотропы — группа лекарственных средств, улучшающих состояние нервной ткани. Современные препараты могут использоваться в раннем возрасте;
- препараты, улучшающие обменные процессы в миокарде. Позволяют предупредить развитие миокардита и его осложнений;
- антибактериальные препараты используются при выявлении у ребенка сопутствующей бактериальной инфекции, например, бронхита или пневмонии;
- успокоительные медикаменты на растительной основе;
- лекарственные комплексы, содержащие коллаген и его предшественники, а также витамин С.
При выборе медикаментозного лечения следует учитывать имеющиеся у ребенка нарушения работы внутренних органов. Все препараты имеют определенные показания и противопоказания к назначению. Их несоблюдение может стать причиной прогрессирования расстройств или развития побочных эффектов.
Изменение рациона питания
Клинические рекомендации по терапии дисплазии в детском возрасте указывают, что ребенок с заболеванием нуждается в специальной диете. Она основана на повышении потребления продуктов, содержащих коллаген, или веществ, стимулирующих его образование в организме.
К коллагенсодержащим продуктам относят все виды нежирного мяса, морскую рыбу и водоросли. Стимулируют образование белка соединительной ткани продукты, богатые витамином С. Например, ягоды, соя, печень животных и птиц, бананы и пр.
Диета — важная часть терапии заболевания. Родителям необходимо проконсультироваться по ее поводу с педиатром и диетологом.
Также из рациона питания устраняют фастфуд, жирную, соленую и острую пищу. Употребление кондитерских и хлебобулочных изделий ограничено.
Хирургические вмешательства
Инвалидность детей с дисплазией связана с изменениями в опорно-двигательном аппарате и сердечно-сосудистой системе. Заболевание приводит к патологиям артерий и вен, деформации позвоночника, грудной клетки и другим негативным изменениям в организме. Для их коррекции врачи проводят оперативные вмешательства, направленные на устранение дефектов. Ребенок при этом нуждается в госпитализации в профильную больницу. Операции проводят после обследования пациента и определения показаний к ним.
Системная дисплазия соединительной ткани в детском возрасте характеризуется множественными клиническими проявлениями — от изменения длины пальцев до тяжелых пороков сердечно-сосудистой системы. Прогноз при своевременном выявлении заболевания благоприятный.
Врачи обеспечивают комплексное лечение, которое тормозит развитие патологических процессов в организме и позволяет предупредить развитие осложнений. Если родители долгое время занимаются самолечением и не обращаются за медицинской помощью, то заболевание может стать причиной гибели ребенка или его инвалидности.
Читайте в следующей статье: акклиматизация у детей











Недифференцированные дисплазии соединительной ткани (проект клинических рекомендаций)
Публикуемый проект второго пересмотра клинических рекомендаций по ведению пациентов с недифференцированными дисплазиями соединительной ткани продиктован наличием обоснованных дополнений/замечаний к ранее утвержденным (в 2018 г.) клиническим рекомендациям.
Дисплазия (dysplasia; греч. dys- + plasis формирование, образование; син. дисгенезия) – неправильное развитие тканей и органов независимо от времени и причины их возникновения [1]. Новые и узко направленные профессиональные термины в настоящих клинических рекомендациях не используются.
1.1. Определение
Недифференцированные дисплазии соединительной ткани (НДСТ; код по МКБ-10 – М35.8) – это генетически детерминированные состояния, характеризующиеся дефектами волокнистых структур и основного вещества соединительной ткани, приводящие к нарушению формообразования органов и систем, имеющие прогредиентное течение, определяющие особенности ассоциированной патологии, а также фармакокинетики и фармакодинамики лекарственных средств [2–4].
Комментарии: генетический дефект может проявляться в любом возрасте в соответствии с временными закономерностями генной экспрессии. Реализация генетических детерминант либо в наибольшей степени определяется внешними условиями, как в случае недифференцированных дисплазий соединительной ткани (несиндромных формах дисплазии соединительной ткани, неспецифических нарушений соединительной ткани), либо мало зависит от внешних условий, как в случае наследственных нарушений соединительной ткани (дифференцированной дисплазии соединительной ткани, синдромных форм дисплазии соединительной ткани) [2–7].
1.2. Этиология и патогенез
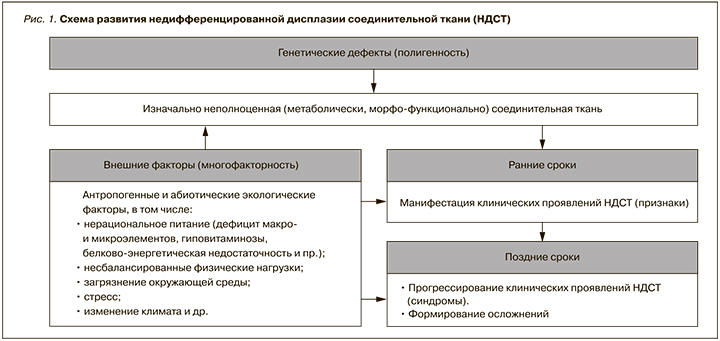
В основе развития дисплазий соединительной ткани (ДСТ) лежат мутации генов, ответственных за синтез/катаболизм структурных белков соединительной ткани или ферментов, участвующих в этих процессах [2–5] (рис. 1).
1.3. Эпидемиология
Распространенность НДСТ – 1:5. Отдельные внешние проявления дисморфогенеза соединительной ткани среди молодых – 85,4% [2–4].
Критическим периодом проявлений НДСТ является подростковый возраст, когда объем соединительной ткани увеличивается пропорционально росту и развитию организма. Как правило, у абсолютного большинства пациентов с НДСТ в возрасте старше 35 лет основную проблему составляют осложнения клинических синдромов, определяющие инвалидизацию пациентов и летальные потери в группе [2–4, 8, 9].
1.4. Кодирование по МКБ-10
1.5. Классификация
В практической работе используется Международная классификация болезней 10-го пересмотра (МКБ-10) [10]. В научных исследованиях можно пользоваться классификацией, предложенной Нью-Йоркской ассоциацией кардиологов, с выделением в нозологическую форму соединительнотканной дисплазии сердца, а также каталогом генов и генетических нарушений человека Mendelian Inheritance in Man (MIM), созданном и редактируемом McKusick V.A. et al., в который вошли такие состояния, как MASS syndrome (Mitral valve prolapse, Aortic root diameter at upper limits of normal for body size, Stretch marks of the skin, Skeletal conditions similar to Marfan syndrome, MIM 604308), Mitral valve prolapse, familial (MIM 157700), Mitral valve prolapse, myxomatous 2, 3 (MIM 607829,610840) и ряд других [2–6].
1.6. Клиническая картина
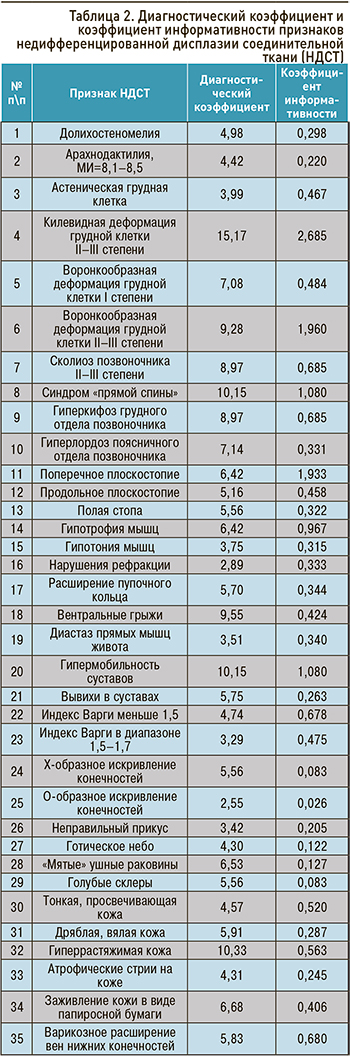
Клинические проявления НДСТ во многом связаны с ведущим клиническим синдромом, затрагивающим ту или иную систему организма (табл. 1). На сегодняшний день выделено 28 синдромов при НДСТ [3].
2.1. Критерии установления диагноза НДСТ
Генетические дефекты могут проявиться в любом возрасте, и чем раньше они появляются, тем более выражена клиническая картина заболевания и тяжелее прогноз. В процессе роста и развития организма накапливаются дефекты в системе CТ: белках внеклеточного матрикса, ферментах, клетках. Возраст появления клинических признаков различных ННСТ зависит от временных закономерностей генной экспрессии, пенетрантности генов и факторов внешней среды.
Известно, что в раннем детском возрасте диагностика СМ вызывает особые затруднения, и на основе клинических данных возможна лишь в 56% случаев. С другой стороны, диагностика СГМС по клинической картине ГМС просто невозможна у детей и подростков
Сегодня усилия исследователей направлены на поиск методов генной инженерии для коррекции генетических дефектов. Вместе с тем, реальная помощь пациентам находится в сфере практической медицины и заключается в разработке общих мероприятий по диагностике и лечению ННСТ в целом и методов профилактики в терапии отдельных форм.
Общие подходы к терапии ННСТ и, прежде всего ДСТ, должны содержать рекомендации по психологической поддержке, режиму дня и двигательной активности, диетотерапии, физическим методам лечения, медикаментозной метаболической терапии.
Несмотря на низкий уровень доказательности, отсутствие клинических исследований эффективности лечения ДСТ, мнение некоторых экспертов состоит в том, что всем пациентам с признаками ННСТ рекомендуется курсовой прием основных 4 групп препаратов, прямо и опосредованно воздействующих на метаболизм СТ.
- I группа - стимуляторы коллагенообразования: витамин С (аскорбиновая кислота), кроме случаев кальциурии и оксалатно-кальциевой кристаллурии, кальцитрин, стекловидное тело, карнитин, солкосерил в сочетании с витаминами группы В (В1, В2, фолиевая кислота, В6) и микроэлементами Cu2+, Zn2+, Mg2+, Мn2+ и др. Среди препаратов 1 группы заслуживает специального упоминания магниевая соль оротовой кислоты, с успехом использующаяся для лечения различных проявлений ДСТ .
- II группа - корректоры нарушения синтеза и катаболизма ГАГ: хондропротекторы из класса хондроитинсульфатов и ГАГ - глюкозаминосульфат, БАДы, содержащие ГАГ.
- III группа - стабилизаторы минерального обмена: L-кальциферол (витамин D2), кальций-D3.
- IV группа - корректоры биоэнергетического состояния организма: препараты, содержащие фосфорные соединения, и БАДы с комплексами эссенциальных аминокислот.
Кроме общих мер по улучшению метаболизма СТ, следует проводить лечебные и профилактические мероприятия, в зависимости от особенностей течения и характера осложнений отдельных форм ННСТ, а также основных сердечно-сосудистых синдромов, сопровождающих ННСТ.
Приложение 1. Терминология
· Акрогерия- (acrogeria; акро- + греч. geron старик; син.: Готтрона акрогерия, Готтрона синдром) наследственная болезнь, характеризующаяся врожденной атрофией кожи конечностей, наиболее выраженной на кистях и стопах, в сочетании с их гипоплазией; наследуется по аутосомно-рецессивному типу.
· Апикальные буллы — располагающийся субплеврально в верхних долях легких эмфизематозный участок, превышающий в диаметре 1 см.
· Арахнодактилия - (arachnodactylia; арахно- +- греч. daktylos палец; паучья кисть).
· Варикоцеле- варикозное расширение яичка и семенного канатика.
· Долихосигма- dolichosigma; греч. Dolichos длинный + анат. [colon] sigmoideum сигмовидная ободочная кишка) - аномалия развития сигмовидной кишки, характеризующаяся ее удлинением.
· Долихостеномелия (dolichostenomelia; долихо - длинный + греч. Stenos –узкий + melos часть тела, конечность) - непропорционально длинные конечности. Диагностируется при измерении длины сегментов туловища.
· Долихоцефалия(dolichocephalia; долихо- + греч. kephale голова; син.: длинноголовость, долихокефалия).
· Миоз(miosis: греч. meiosis –уменьшение, убыль) - сужение зрачка (диаметр менее 2.5 мм).
· Ретрогнатия- (retrognathia; ретро- + греч. gnathos челюсть) сдвиг верхней челюсти назад при ее нормальных размерах.
· Рубцы атрофические - плоские, мягкие, малоподвижные в результате атрофии клетчатки под ними. Кожа рубца истончена, не выступает над здоровой кожей.
· Спондилолистез- смещение позвонков относительно друг друга.
· Стрии атрофические - растяжки на коже, возникшие в результате истончения и утраты эластичности ее внутренних слоев и разрушения пучков коллагеновых волокон.
· Трабекула нормальная - мышечный тяж, плотно примыкающий к эндокарду желудочка.
· Трабекула аномальная — мышечный или фиброзно-мышечный тяж, не плотно примыкающий к эндокарду желудочка или соединяющий стенку желудочка с межжелудочковой перегородкой.
· Трахеобронхиальная дискинезия - повышенная подвижность стенок трахеи и бронхов: расширение при вдохе и сужение просвета на выдохе.
· Трахеобронхомаляция —(tracheobronchomalacia) диффузное или очаговое размягчение хрящей трахеи и бронхов связанное с врожденными морфологическими дефектами хрящевого и соединительнотканного каркаса трахеи и бронхов.
· Трахеобронхомегалия - (синдром Мунье-Куна) представляет собой врожденное чрезмерное расширение трахеи и крупных бронхов.
· Хорда левого желудочка истинная - фиброзный тяж, соединяющий папиллярную мышцу со створкой клапана.
· Хорда левого желудочка ложная - фиброзно-мышечный или фиброзный тяж, соединяющий папиллярные мышцы между собой или со стенкой желудочка, или межжелудочковой перегородкой.
· Эпикантус- поперечная кожная складка около внутреннего угла глаза, обычно двусторонняя.
· Энофтальм - (от греч. en - в. внутри и ophthalmos -глаз), глубокое положение глазного яблока в глазнице.
1. НАРУШЕНИЯ АНАТОМИЧЕСКОГО СТРОЕНИЯ ОРГАНОВ В РЕЗУЛЬТАТЕ ПАТОЛОГИИ РАЗВИТИЯ СОЕДИНИТЕЛЬНОЙ ТКАНИ, СОПРОВОЖДАЮЩИЕСЯ КЛИНИЧЕСКИ ЗНАЧИМЫМ НАРУШЕНИЕМ ФУНКЦИИ ОРГАНОВ, ОТНОСЯТ К
1) малым аномалиям развитиям
2) порокам развития органов
3) дисплазиям соединительной ткани
4) гипермобильности суставов
2. КЛАССИФИКАЦИЯ ДСТ
1) основана на типе наследования
2) в настоящее время не разработана
3) учитывает только клиническую симптоматику
4) выделяет дифференцированные и недифференцированные ДСТ
3. НАИБОЛЕЕ ЧАСТО ВСТРЕЧАЮЩИЕСЯ КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ ДСТ
1) гипермобильность суставов, ПМК, вегетососудистая дистония
2) аневризмы сосудов, птозы внутренних органов, арахнодактилия
3) гипермобильность суставов, рецидивирующие гематомы, астенический тип телосложения
4) варикозное расширение вен нижних конечностей, сколиотическая деформация грудной клетки, пролапс трикуспидального клапана
4. ТИП НАСЛЕДОВАНИЯ СИНДРОМА МАРФАНА
3) Х-сцепленный с полом
5. ОСНОВНЫМ КЛИНИЧЕСКИМ ПРОЯВЛЕНИЕМ СИНДРОМА ЭЛЕРСА-ДАНЛО
1) гипермобильность суставов
2) гиперрастяжимость кожи
3) астеническое телосложение и паукообразные пальцы
4) патология скелетной системы и органа зрения
6. ЛАБОРАТОНЫЙ ТЕСТ, ПОДТВЕРЖДАЮЩИЙ НАЛИЧИЕ СГМС НАПРАВЛЕН НА ОПРЕДЕЛЕНИЕ
1) уровня в крови гидроксипролина
2) содержания в суточной моче оксипролина или гидроксипролина
3) уровня тенасцина X сыворотки крови
4) содержания в суточной моче гликозаминогликанов
7. НЕПРОПОРЦИОНАЛЬНО ДЛИННЫЕ КОНЕЧНОСТИ НАЗЫВАЮТ
8. ЭПИКАНТУСОМ НАЗЫВАЮТ
1) глубокое положение глазного яблока в глазнице
2) поперечная кожная складка около внутреннего угла глаза, обычно двусторонняя
3) монголоидный разрез глаз
4) дополнительная продольная кожная складка над верхним веком
1. Молодой человек 16 лет в ходе призывной комиссии в военкомате проходит медицинский осмотр. Жалоб не предъявляет. Из анамнеза: длительно состоит на учете окулиста по поводу подвывиха хрусталика. При осмотре: рост 198 см, вес 63 кг, кожные покровы обычной окраски, длина размаха рук 192 см, грудная клетка узкая и плоская, эпигастральный угол менее 90 ˚, ход ребер косо нисходящий, межреберные промежутки увеличены, сколиоз грудного отдела позвоночника, плоскостопие, признаки гипермобильности суставов. При аускультации сердца: ЧСС=80 в мин, 1 тон ˃ 2 тона, на верхушке слабый систолический шум убывающего характера, проводящийся в подмышечную область; при аускультации легких без особенностей. Живот пальпаторно мягкий, безболезненный.
1) Предварительный диагноз?
2) Дальнейшая тактика в плане обследования и лечения?
2. Мужчина 24 лет, направлен на консультацию генетика. Жалобы на рецидивирующие гематомы, снижение зрения. Осмотр: рост 180 см, вес 74 кг, кожные покровы обычной окраски, при образовании кожной складки в подключичной области её величина более 5 см; на передней брюшной стенке широкий атрофичный рубец после аппендэктомии; Ps=ЧСС=72 в мин, АД 120 и 80 мм рт ст, при осмотре и аускультации сердечно-сосудистой и бронхо-легочной систем без особенностей; осмотр кардиолога: ПМК 1 ст, НЦД по кардиальному типу; хирург: сколиоз 1 ст, продольное плоскостопие 2 ст; пульмонолог: бронхоэктатическая болезнь; окулист: миопия 2 ст.
1) Предварительный диагноз?
2) Дополнительное обследование и рекомендации?
3. Студентка 2 курса СГМУ, обратилась к терапевту с жалобами на повышенную утомляемость, ощущение учащенного сердцебиения при физической нагрузке, снижение зрения в течение года. Из анамнеза известно, что с детства занимается легкой атлетикой. При осмотре: рост 182 см, вес 68 кг, астеническое телосложение, признаки гипермобильности суставов; арахнодактилия; Ps=ЧСС=58 в мин, АД 105 и 70 мм рт ст, при осмотре и аускультации сердечно-сосудистой и бронхо-легочной систем без особенностей; живот при пальпации мягкий, безболезненный. ЭКГ показатели в норме.
2) Необходимость молекулярно-генетического исследования?
ОНКОГЕНЕТИКА
Онкогенетика (онко- + генетика) – раздел онкологии, изучающий роль генетических факторов в этиологии и патогенезе опухолей.
Злокачественные новообразования относятся к мультифакториальным заболеваниям, в развитии которых играют роль как генетические факторы, так и факторы окружающей среды, называемые канцерогенными.
В 1869 г. французский хирург Брока впервые описал родословную семьи своей жены, в которой 10 женщин из 24 умерли от рака молочной железы, таким образом, впервые указав на роль наследственности, как одного из механизмов канцерогенеза.
В начале XXв. была предложена мутационная теория рака, в которой подчеркивалась роль генных и хромосомных соматических мутаций в этиологии рака.
В последние годы все большую популярность приобретает вирусо-генетическая теория рака, согласно которой генетический материал онкогенных вирусов встраивается в хромосому клетки. Такое изменение генома, а вернее, его отдельных локусов, нарушает биохимический механизм клеток, они приобретают автономность и начинают усиленно делиться.
Злокачественные новообразования относятся к группе генетических соматических болезней (генетических болезней соматических клеток). Злокачественные клетки всегда имеют мутационные изменения на генном, хромосомном или геномном уровне.
Мутации, определяющие развитие опухоли, могут быть:
- герминативными (существуют уже в гаметах и, следовательно, во всех клетках организма, передаются от родителей потомству из поколения в поколение);
- соматическими (возникают в соматических клетках в результате спонтанного или индуцированного мутационного процесса, не передаются потомству).
Большинство злокачественных новообразований развивается в результате случайной мутации в одной единственной клетке (соматической), которая в последующем при делении дает начало опухоли. Однако около 10-15% случаев злокачественных новообразований имеет наследственный характер, то есть мутация, предрасполагающая к заболеванию раком, передаётся из поколения в поколение.
Основные генетические механизмы, запускающие процесс канцерогенеза, связаны с мутацией генов двух групп семейств – протоонкогенов и антионкогенов (генов-супрессоров опухолевого роста).
Протоонкогены играют ключевую роль в пролиферации и дифференцировке клеток, репарации ДНК, работе клеточных рецепторов. В организме протоонкогены могут трансформироваться в онкогены и запускать генетический процесс озлокачествления. Существует несколько механизмов такой трансформации. Например, мутации в кодирующих последовательностях протоонкогенов могут вызвать их активацию, что приведет к экспрессии онкогенных белков и накоплению в клетке онкогенных продуктов. Протоонкогены на протяжении всей жизни человека находятся в функционально неактивном состоянии либо очень слабоактивны. Мутации в протоонкогенах в большинстве случаев возникают в соматических клетках под действием мутагенных факторов внешней среды (промышленные и сельскохозяйственные яды, табачный дым, вирусы). В связи с этим развитие большинства распространенных злокачественных новообразований у взрослых связанно с мутациями именно в этих генах (спонтанный мутационный процесс). Однако встречаются и наследственные формы заболеваний (пигментная ксеродерма, атаксия-телеангиэктазия Луи—Бар, синдром Блума). Если мутации происходят в разных частях одного гена, это может приводить к различным болезням. Например, мутации в разных частях RET-онкогена ведут к 4 клинически разным наследственным заболеваниям: двум формам множественной эндокринной неоплазии, семейной медуллярной тиреоидной карциноме, семейной болезни Гиршпрунга.
Антионкогены, напротив, ответственны за подавление роста опухолей, в связи с чем также называются генами-супрессорами опухолевого роста. Это аутосомно-доминантные гены. Их функция – подавлять нецелесообразную пролиферацию клеток. Дефекты этих генов приводят к прогрессии, а восстановление функции – к существенному замедлению пролиферации или даже обратному развитию опухоли. В настоящее время идентифицировано более 20 супрессорных генов, мутации в которых приводят к развитию опухолей. Несмотря на то, что мутации в этих генах наследуются аутосомно-доминантно, механизмы канцерогенеза реализуются лишь в гомозиготных или гемизиготных по мутантному аллелю клетках. Это связано с определенным типом мутации в антионкогенах, приводящей к потере гетерозиготности. При гетерозиготном состоянии аллеля (Аа) развитие опухоли не наблюдается.
Некоторые факторы внешней среды способны приводить как к активации протоонкогенов, так и к инактивации генов-супрессоров, т.е. обеспечивать реализацию обоих механизмов развития опухолей. В качестве примера может служить возникновение рака легких при нарушениях биотрансформации одного из компонентов табачного дыма — бензопирена. Один из его метаболитов способен связываться с ДНК и вызывать активацию протоонкогенов или инактивацию генов-супрессоров опухолевого роста, что и приводит к развитию злокачественных новообразований.
Дата добавления: 2018-06-01 ; просмотров: 248 ;
Читайте также:
