
Мышечная дистрофия лечение стволовыми клетками
16 февраля 2016
- 3800
- 3,1
- 0
- 6
Принцип генной терапии миодистрофии Дюшенна/Беккера. Миодистрофию Дюшенна (МДД) вызывают мутации гена дистрофина (DMD), приводящие к сдвигу рамки считывания, а более мягкую миодистрофию Беккера (МДБ) — мутации без смещения рамки считывания. Лечения этой болезни пока нет. Генная терапия поможет улучшить или даже восстановить функции мышц.
![]()
Анна Петренко

![]()
Антон Чугунов![]()
Ольга Волкова


- CRISPR/CAS
- Генная инженерия
- Медицина
Мышечная дистрофия Дюшенна — тяжелейшее Х-связанное заболевание, эффективного лечения которого до сих пор нет. В одном из последних номеров Science вышли целых три статьи об успешном тестировании на мышиных моделях технологии CRISPR/Cas9 для лечения этой болезни. Может быть, у этого подхода есть шанс добраться и до клиник?
Мышечная дистрофия Дюшенна, от которой страдает один из 3600-5000 новорожденных мальчиков, вызывается отсутствием дистрофина — белка, который соединяет цитоскелет и внеклеточный матрикс в мышечном волокне и обеспечивает его стабильность при сокращении (рис. 1). Из-за мутаций гена DMD рамка считывания при трансляции его мРНК сдвигается, и синтез белка преждевременно прекращается. Врожденная болезнь очень быстро прогрессирует: ее диагностируют в возрасте около четырех лет, а к 10 годам ребенку обычно уже нужна инвалидная коляска. Это происходит потому, что без дистрофина волокна повреждаются, и как только регенеративная способность мышечных волокон исчерпывается, они заменяются фиброзной и жировой тканями [1]. Как показывают исследования, когнитивные функции у ребенка тоже могут быть нарушены [2]. Больше 30 лет с таким заболеванием, как правило, не живут, а смерть наступает от сердечных и респираторных осложнений. Более мягкий вид миодистрофии, связанной с геном DMD, — это мышечная дистрофия Беккера, когда мутации не приводят к смещению рамки считывания [3].
Дистрофин находится на внутриклеточной поверхности сарколеммы вдоль всей длины мышечных волокон и входит в состав дистрофин-ассоциированного гликопротеинового комплекса (ДАГК, DGC). Он связывается одним концом с F-актином цитоскелета, а другим — с β-дистрогликаном, что стабилизирует волокна во время сокращения. Ген дистрофина — один из самых длинных у человека.
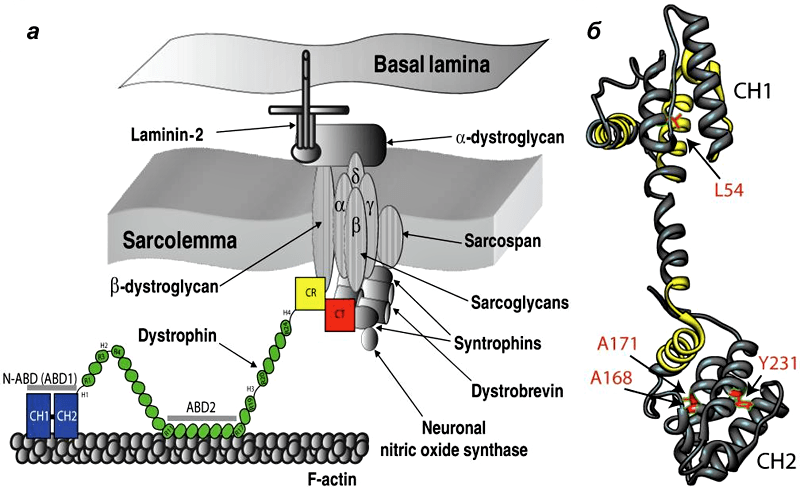
Рисунок 1. Мутации в дистрофине — причина развития миодистрофии Дюшенна. а - Дистрофин связывается с актиновыми филаментами (часть цитоскелета) через домены N-ABD и ABD2) и с ДАГК через домены CR и CT. б — Кристаллическая структура N-ABD дистрофина. Зоны связывания с актином показаны желтым, четыре хорошо изученных мутации, вызывающих заболевание, — красным.
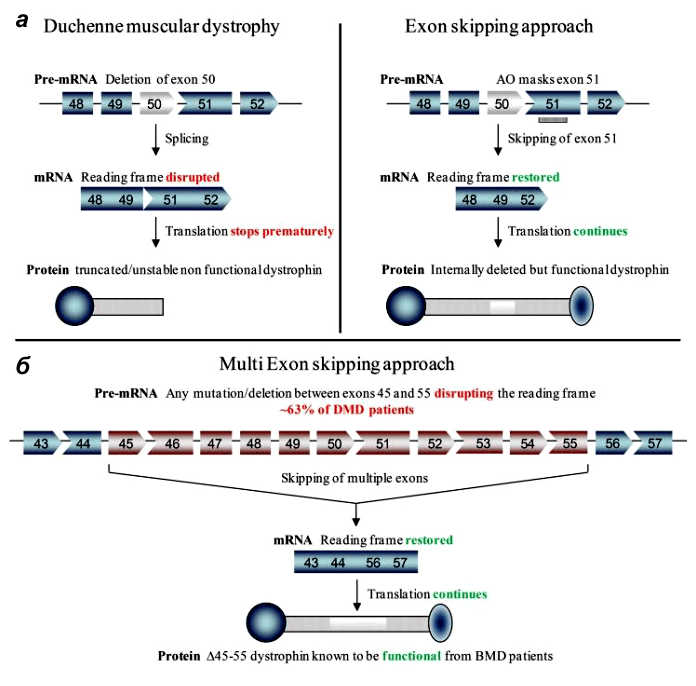
У стратегии удаления экзонов есть даже преимущества перед воссозданием полной длины гена: ее проще разработать, чем восстановить индивидуальные делеции каждого пациента [7].
Конкурирующие лаборатории: кто первым воплотит технологию в терапию для человека?
Ученые трех лабораторий успешно применили технологию пропуска экзонов in vivo на стандартном объекте — мышах — и показали, что их метод помогает восстановить рамку считывания и частично восстановить синтез дистрофина. Поскольку даже невысокий его уровень (3–15% от нормального) приносит терапевтическую пользу, результаты работ можно назвать успешными.
Группа Эрика Олсона уже не в первый раз использует метод CRISPR/Cas9 в своих работах по мышечной дистрофии Дюшенна. В 2014 году ученые исправили мутацию в зародышевой линии мышей и предотвратили развитие болезни. Однако, поскольку пренатальное редактирование генома на человеческих эмбрионах (пока?) запрещено, исследователям пришлось придумать способ постнатального применения технологии.
Группа Эми Уаджерс провела во многом похожий эксперимент [8]. После множества подготовительных этапов работы по редактированию генома и пропуску экзона на клетках и животных их опыт тоже увенчался успехом: программируемые CRISPR-комплексы в составе аденоассоциированного вируса (AAV) были доставлены с помощью локального и системного введения к дифференцированным скелетным волокнам, кардиомиоцитам и сателлитным мышечным клеткам новорожденных и взрослых мышей. Если редактирование направлено только на мышечные волокна, то эффект со временем может сойти на нет. Однако, как отмечает Уаджерс, редактирование генов в сателлитных клетках может обеспечить гораздо более длительный результат. Оно способно привести к созданию пула регенеративных клеток, несущих отредактированный ген дистрофина, и в результате обычной репарации мышц отредактированный ген окажется и в мышечных волокнах.
Терапия миодистрофии Дюшенна: старые и новые подходы
По словам Олсона, главное отличие новой стратегии с использованием вектора, вмещающего в себя компоненты для редактирования генома, от других терапевтических методов в том, что она устраняет причину болезни. А какие еще подходы разрабатывают ученые?
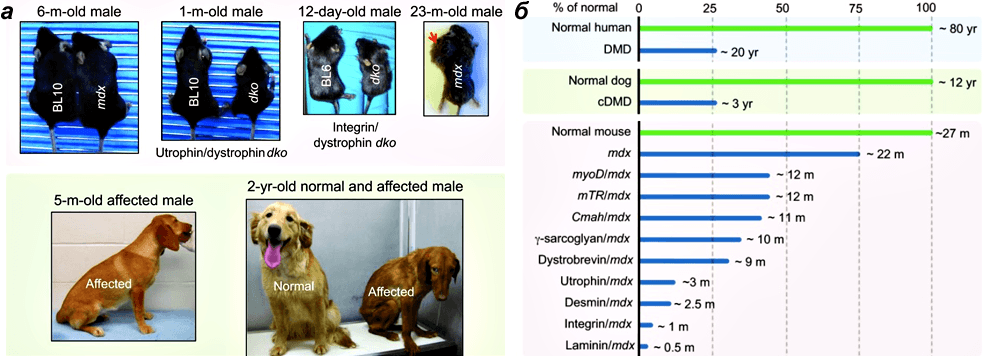
Рисунок 3. Животные модели миодистрофии Дюшенна. а — Проявления миодистрофии Дюшенна у мышей и собак. Вверху: у мышей mdx симптомы проявляются только в старости, и они склонны к образованию рабдомиосарком — опухолей мышечного происхождения. Размер мышей с нокаутами генов атрофина/дистрофина и интегрина/дистрофина значительно меньше, чем их ровесников дикого типа (BL10 и BL6). Внизу: проявления болезни у пятимесячной больной собаки. Различия между здоровой и больной двухлетними собаками. б — Сравнение продолжительности жизни здоровых и больных людей, собак и различных линий мышей.
Один из многообещающих подходов — это клеточная терапия. Хотя опыты с внутримышечной инъекцией миобластов от здоровых доноров провалились, технологии с использованием стволовых клеток и индуцированных плюрипотентных стволовых клеток (ИПСК) пока успешно испытываются на моделях не только миодистрофии Дюшенна, но и болезни Альцгеймера, Паркинсона, Хантингтона, спинальной мышечной атрофии, бокового амиотрофического склероза, аутизма и шизофрении [14–16]. Например, в 2013 году исследователи из Бостонской детской больницы (Boston Children’s Hospital’s Stem Cell Program) с помощью смеси трех малых молекул (форсколина, основного фактора роста фибробластов bFGF и ингибитора гликогенсинтазы киназы-3) перепрограммировали ИПСК из кожи пациентов с миодистрофией Дюшенна в мышечные клетки, которые затем успешно прижились у мышей. Сейчас из ИПСК получены кардиомиобласты и нейроны [2].
Другие исследования показывают, что восстановление нормального уровня синтеза оксида азота (NO), который снижается у больных из-за нарушения активности NO-синтазы (nNOS), ослабляет воспаление, повышает активность собственных стволовых клеток и реконструирует морфологию и функции скелетных мышц [3].
Уже в фазе II клинических испытаний находится препарат Givinostat — ингибитор гистондеацетилаз, который замедляет прогрессирование болезни в мышиной модели.
Такой массированный экспериментальный удар по миодистрофии Дюшенна вселяет надежду. Станет ли технология CRISPR/Cas9 ведущей в разработке терапии, которую смогут принять на вооружение клиницисты? Возможно, не за горами и публикации похожих работ по другим заболеваниям, где нужно избавиться от мутаций в одном-единственном гене? Это мы узнаем из ближайших выпусков Science (а также других почетных журналов).
ДЛЯ ТОГО ЧТОБЫ СКАЧАТЬ СТАТЬЮ В ФОРМАТЕ PDF ВАМ НЕОБХОДИМО АВТОРИЗОВАТЬСЯ, ЛИБО ЗАРЕГИСТРИРОВАТЬСЯ
В обзоре обсуждаются современные подходы к лечению мышечных дистрофий - гетерогенной группы нервномышечных заболеваний, которые проявляются в виде прогрессирующей слабости мышц, а также их частичной потери, что во многих случаях приводит к смерти. Эффективных медикаментозных способов лечения дистрофий в настоящее время нет. Однако существует несколько исследуемых терапевтических вариантов лечения мышечных дистрофий, включающих генную терапию и трансплантацию миогенных клеток-предшественников - клеточную терапию. В статье обсуждаются достижения и трудности на пути внедрения генной терапии в клиническую практику лечения мышечных дистрофий. Особое внимание уделено клеточной терапии, перспективному направлению регенеративной медицины, дающему надежду на излечение многих ранее не излечимых как наследственных, так и приобретенных заболеваний. Обсуждаются потенциальные источники соматических и эмбриональных стволовых/прогениторных клеток человека, которые могут использоваться как в качестве объектов приложения генно-инженерных методов, так и для клеточной терапии мышечных дистрофий. Обсуждаются проблемы, стоящие на пути успешного внедрения в клиническую практику лечения мышечных дистрофий стволовых/проге-ниторных клеток человека.
Введение
Мышечная дистрофия - это группа наследственных заболеваний, которые характеризуются прогрессирующей мышечной слабостью и потерей мышечных тканей. В настоящее время все дистрофии считаются заболеваниями неизлечимыми. В некоторых случаях болезнь приводит к смерти. Мышечные дистрофии различаются по группам поврежденных мышц, возрасту, при котором начинается болезнь, и скорости ее прогрессирования. Молекулярной причиной, лежащей в основе многих дистрофий, являются мутации в генах, кодирующих компоненты дистрофин-гликопротеинового комплекса, который связывает миофибрильный цитоскелет с внеклеточным матриксом 1.
Наиболее часто встречается и наиболее тяжело протекает мышечная дистрофия Дюшенна (МДД). МДД поражает мальчиков с частотой, равной 1 на 3500. Она проявляется в 1-2-летнем возрасте и характеризуется потерей независимого передвижения уже к 10 годам. Больные МДД, как правило, умирают в возрасте 15-25 лет от сердечной или легочной недостаточности вследствие атрофии дыхательных и сердечной мышц. Сходными симптомами и причиной возникновения характеризуется мышечная дистрофия Беккера (МДБ), однако протекает это заболевание в менее тяжелой форме. Обе болезни являются следствием мутаций в гене дистрофина, который локализован на коротком плече Х-хромосомы и является самым большим из известных генов человека. Он содержит 79 экзонов и составляет приблизительно 0,1 % всего генома человека 5. Этот ген кодирует мышечный белок дистрофин с молекулярным весом 427 кДа, который является основным структурным белком, стабилизирующим сарколемму мышечного волокна. Своим N-концом он связывается с актином - главным компонентом внутриклеточного цитоскелета, а С-концом - с группой дистрофин ассоциированных дистро- и саркогликанов -белков сарколеммы, и через них - с основным белком внеклеточного матрикса - ламинином (рис. 1).
Мутации в гене дистрофина приводят к полному отсутствию или сильному снижению уровня дистрофина в случае МДД, либо к образованию частично функционального дист-рофина в случае МДБ. Отсутствие или функциональная недостаточность этого белка приводит к нарушению целостности клеточной мембраны и является причиной запуска процессов дегенерации мышечного волокна. Результатом этого процесса является повышение проницаемости мембран и возникновение в них физических разрывов, что приводит к выходу ферментов из мышц в сыворотку крови (повышение в сыворотке крови активности креатинкиназы является биохимическим маркером МДД и МДБ). На начальных стадиях заболевания дегенерация мышечных волокон компенсируется активной регенерацией фибрилл, благодаря делению и слиянию мышечных сателлитных клеток. Однако с возрастом этот процесс становится менее эффективным, что приводит к прогрессирующей мышечной слабости [7].
В настоящее время при отсутствии эффективных медикаментозных способов возлагаются большие надежды на использование для лечения мышечных дистрофий методов генной и клеточной терапий. Задачей этих способов лечения является восстановление синтеза дистрофина в мышечных клетках, что достигается либо путем встраивания в клетки нормального гена дистрофина, либо коррекцией мутаций в самом гене или в иРНК [8].
Генная терапия мышечных дистрофий
Основное различие генной и клеточной терапий заключается в способе доставки нормального гена дистрофина в мышечную клетку. Генно-инженерные подходы доставки гена используют, как правило, вирусные векторы. В экспериментах на мышах была продемонстрирована достаточно эффективная и долговременная трансфекция скелетных и сердечной мышц, а также мышц диафрагмы после введения аденовирусных, ретровирусных или лентивирусных векторов, доставляющих функциональный ген дистрофина или минидистрофина 10.
Вместе с тем, использование вирусных носителей, особенно в экспериментах in vivo, наталкивается на существенные методические трудности. К ним, например, относятся недостаточная пакующая способность ретровирусов и необходимость наличия клеток-хелперов. Наибольшим препятствием использования вирусных векторов для доставки генетических конструкций является выраженный иммунный ответ реципиента на вирусные антигены. Несмотря на огромный объем работ по модификации генома вируса-носителя и сокращению его размера до минимально возможного, иммунный ответ, тем не менее, сохраняется и делает бессмысленным повторное введение генно-инженерных конструкций. В результате оказывается невозможно достичь такого уровня трансфекции, при котором наступает эффект лечения. Большинство исследователей полагает, что достижение терапевтического эффекта возможно при успешной трансфекции не менее 20% [по последним данным - даже 40%) всех мышечных волокон, включая мышцы сердца и диафрагмы. При этом основными критериями эффективности трансфекции являются появление дистрофин-положительных мышечных волокон, нормализация уровня креатинкиназы в сыворотке крови, изменение физиологических параметров [силы мышц и др.).
Существуют также невирусные способы доставки к ДНК гена дистрофина, которые включают в себя баллистическую трансфекцию, методы электропорации [электрошока), введение генетических конструкций в составе липосом, либо упакованных с помощью олигопептидов, молекулярных конъюгатов или полимерных носителей [9, 13, 14]. Эти способы не вызывают такого иммунного ответа, как вирусные векторы, однако способность к трансформации у большинства из них ниже.
Несмотря на довольно интенсивные исследования на животных моделях, до настоящего времени клинические испытания генно-инженерных методов лечения дистрофий на больных людях практически не проводились.
Следует также упомянуть о существовании еще одного подхода к лечению дистрофий - активации синтеза в мышечных клетках репрессированного в процессе онтогенеза аутосомного гомолога гена дистрофина - гена утрофина [15]. Известно, что в эмбриогенезе человека приблизительно до семи недель развития дистрофин не экспрессируется, и его функцию в мышцах выполняет белок утрофин [16]. Между 7 и 19 неделями развития экспрессируются оба белка, после чего происходит замещение мышечного утрофина на дистрофин. Эти белки похожи своими N- и С-концевыми доменами, играющими решающую роль в функции дистрофи-на, тогда как функционально малозначимый центральный rod domain присутствует в утрофине в сильно укороченном виде. Если достичь дерепрессии гена утрофина у больных мышечной дистрофией, то продукт его экспрессии белок утрофин мог бы компенсировать недостаток дистрофина в мышцах [17]. В настоящее время получены данные, свидетельствующие о том, что in vivo трансфекция mdx мышей геном утрофина приводит к экспрессии этого белка в скелетных мышцах и диафрагме 20. Эти результаты указывают на принципиальную возможность коррекции недостатка дистрофина в мышечных волокнах с помощью утрофина.
Клеточная терапия мышечных дистрофий
Перспективным направлением клеточной терапии мышечных дистрофий представляется трансплантация как стволовых клеток [СК), способных к самовозобновлению и дифференциации во множество клеточных линий организма, так и миогенных прогениторных клеток. При этом рассматриваются возможности использования для трансплантации как взрослых, так и эмбриональных стволовых/прогенитор-ных клеток.
Стволовые клетки костного мозга. В КМ, основываясь на адгезивных свойствах in vitro, различают два типа СК: адгезивные стромальные мезенхимальные СК и неадгезивные гемопоэтические СК. Мезенхимальные СК могут дифференцироваться в клетки кости, хряща, жировой ткани и мышц как in vitro, так и in vivo. Миогенные клетки могут быть получены in vitro при действии на адгезивные мезенхимальные клетки 5-азацитидина (5-azacytidine), который активирует мышечный переключатель MyoD [35]. Субпопуляция мезенхимальных СК также была идентифицирована в мо-нонуклеарных клетках КМ - это CD45+/glycophorin+ клетки [36]. После ex vivo экспансии эти мультипотентные взрослые прогениторные клетки могут быть трансплантированы в необлученный организм мышей, где они приживаются и развиваются в гемопоэтические линии, эпителиальные клетки, клетки печени, легких, кишечного эпителия. При введении в раннюю бластоцисту эти клетки дифференцировались в мышечные. И хотя их способность становиться предшественниками скелетной мышцы при внутривенном введении продемонстрирована не была, свойство этих мультипотент-ных взрослых прогениторных клеток активно пролиферировать без очевидного старения и потери мультипотенции делает их привлекательным кандидатом для клеточной терапии.
Более перспективными для клеточной терапии мышечных дистрофий являются неадгезивные гемопоэтические СК, которые, помимо гемопоэза, участвуют в формировании нервной ткани [37, 38], печени [39], сердечной мышцы [29] и скелетных мышц. Было показано, что эти миогенные прогениторы могут мигрировать и участвовать в регенерации поврежденной мышечной ткани после трансплантации КМ [40]. В этом исследовании также было продемонстрировано, что в репарации поврежденных кардиотоксином мышечных клеток могут участвовать не только клетки КМ, введенные непосредственно в мышцу, но также и введенные внутривенно. В обоих случаях донорские клетки сливались с мышечными волокнами реципиента. Эксперименты по внутривенному введению донорских клеток КМ mdx мышам также продемонстрировали, что трансплантированные клетки попадают из кровеносной системы в мышечную ткань и сливаются с волокнами дистрофических мышц mdx мышей [30, 41, 42]. Однако, во всех этих экспериментах репарация дистрофических мышц трансплантированными клетками КМ никогда не превышала 1 %. Это можно объяснить как слабым захватом клеток КМ, так и неспособностью стволовых клеток КМ адекватно реагировать на клеточные сигналы реципиента.
Несмотря на то, что использование клеток КМ для лечения модели МДД у животных оказалось неэффективным, существует мнение некоторых исследователей, что трансплантация КМ для лечения мышечной дистрофии у людей будет более эффективной. Способность СК КМ человека участвовать в репарации мышечных волокон была продемонстрирована Gussoni с соавторами [43]. Они представили иммуногистохимический и FISH анализ мышц мальчика, которому в возрасте 1 года для лечения тяжелого X-связанного иммунодефицита, диагностированного вместе с МДД, была проведена трансплантация КМ. При этом, в 12 лет МДД имела умеренное протекание, а в мышцах больного через 13 лет после трансплантации было обнаружено присутствие диплоидных донорских ядер [0,5-0,9% мышечных волокон), что указывает на способность экзогенных клеток КМ человека сливаться со скелетными мышечными волокнами реципиента и сохраняться на протяжении, по крайней мере, 13 лет.
Таким образом, несмотря на возможность взрослых ге-мопоэтических СК сливаться с дистрофическими скелетными мышечными волокнами, частота таких случаев слишком низка для того, чтобы быть эффективным способом лечения мышечных дистрофий.
Клетки пуповинной крови. Очень близки по свойствам клеткам КМ клетки пуповинной крови. В их составе также присутствуют как кроветворные, так и мезенхимальные СК [44]. Применение этих клеток лишено морально-этических ограничений, их легко получать, количество СК в пуповинной крови значительно больше, пролиферативный потенциал выше, они не отягощены грузом генетических мутаций, накопленных взрослыми соматическими СК КМ. Клетки пуповинной крови менее иммунологически зрелые. Эксперименты, проведенные на мышах [45], продемонстрировали, что миогенные прогениторы [предшественники) присутствуют в пуповинной крови человека и способны мигрировать и дифференцироваться в миоциты. Однако, как и в случае СК КМ, до сих пор отсутствуют доказательства эффективного терапевтического действия клеток пуповинной крови при их использовании для лечения мышечных дистрофий. Как и для всех остальных потенциальных источников клеток для лечения мышечных дистрофий, существуют методические проблемы, связанные с выделением из пуповинной крови популяции миогенных предшественников и их дальнейшей экспансией в условиях in vitro.
СатК были одним из первых типов клеток, используемых в клеточной терапии мышечных дистрофий. СатК, которые выращиваются in vitro, представляют собой мононуклеар-ные клетки, коммитированные по миогенному пути [миоб-ласты). Эксперименты по внутримышечной трансплантации миобластов, изолированных из мышей дикого типа, mdx мышам продемонстрировали экспрессию дистрофина in vivo [55, 56]. Эти исследования послужили основанием проведения клинических испытаний миобластов с целью лечения МДД 56. Для этого донорские СатК были изолированы в результате биопсии мышц у близких родственников больных детей. Затем эти клетки выращивались в условиях in vitro, после чего 80-100 миллионов донорских миобластов было введено в мышцы детей путем множественных инъекций. Контрлатеральные мышцы были инъецированы как контроль плацебо. Однако, иммуногистохимические и RT-PCR исследования мышц больных, которые проводились на протяжении года, показали, что трансплантация миобластов таким способом была неэффективна. Несмотря на то, что пересаженные миобласты действительно сохранялись и продуцировали дистрофин в мышечных волокнах пациентов с МДД, частота, при которой это встречалось, была очень низкой (
Клинический случай
Пациент в возрасте 20 лет с МДБ. Заболевание началось со слабости в ногах и частых падений во время ходьбы в возрасте 9 лет. Постепенно развивалась сильная боль в икрах, стало сложно подниматься с пола. В 14 лет стал испытывать сложности при подъеме по лестнице. Симптомы носили прогрессирующий характер. Пациент был проконсультирован педиатром и неврологом. На основании повышенного уровня креатинфосфокиназы (КФК) и клинических проявлений была диагностирована мышечная дистрофия. Спустя год пациенту стало сложно справляться с повседневной физической нагрузкой: вставать с пола / кресла, подниматься по лестнице, сохранять равновесие при ходьбе (частые падения). Пациент прошел курс физиотерапии и приема комплекса витаминов, что не принесло результата.
По данным неврологического осмотра выявлена мышечная гипотония и гиперрефлексия с проксимальной мышечной слабостью, скованностью в ахилловых сухожилиях, псевдогипертрофией икроножных, дельтовидных, ягодичных мышц и мышц предплечья. У пациента наблюдалась походка вразвалку с желанием опереться, переразгибание коленей, гиперлордоз, быстрая утомляемость и периодически возникающая боль в груди. Для оценки состояния пациента были использованы различные критерии, которые приведены в Таблице 1.
| Критерии оценки | До медицинского вмешательства | После медицинского вмешательства (спустя 3 месяца) | После медицинского вмешательства (спустя 9 месяцев) |
| Мера функциональной независимости (FIM), баллы | 113 | 113 | 113 |
| Шкала равновесия Берга (BBS), баллы | 37 | 37 | 37 |
| Шкала способности к самостоятельному передвижению North Star (NSAA), баллы | 15 | 15 | 18 |
| Шкала Брука, баллы | 1 | 1 | 1 |
| Шкала Виньоса, баллы | 3 | 3 | 3 |
| Жизненная емкость легких (ЖЕЛ), мл | 1250 | 1750 | 2000 |
| Пиковая скорость выдоха (ПСВ), мл | 290 | 360 | 320 |
По данным лабораторных исследований уровень КФК был повышен (3180 МЕ/л). МРТ мышц выявило диффузную мышечную атрофию и жировое замещение ягодичной, бедренной мышц и мышц предплечья. Электромиография (ЭМГ) выявила полифазные моторные единицы со сниженной длительностью и амплитудой потенциала действия, что служит причиной миопатического процесса. Эхокардиография(ЭхоКГ) с допплерографией и цветным картированием выявила генерализованную гипокинезию, малую сократимость левого желудочка и его диастолическую дисфункцию третьего типа. Фракция выброса левого желудочка составила 25-30%.
Материалы и методы
Пациент был выбран для медицинского вмешательства в соответствии с Хельсинкской Декларацией Всемирной Медицинской Ассоциации (ВМА). Протокол лечения разработан Институциональным комитетом исследования стволовых клеток (Institutional Committee for Stem Cell Research, IC-SCR). Перед госпитализацией пациент подписал добровольное информированное согласие на медицинское вмешательство.
Проведена детальная оценка. Мышцы с показателем функциональной независимости меньше 3 (бицепс, трицепс, квадрицепс, разгибатели спины, ягодичная/брюшная/ передняя большеберцовая мышцы, мышцы задней поверхности бедра) выбраны для внутримышечной трансплантации мезенхимальных стволовых клеток костного мозга (далее – МСККМ). Двигательные точки (проекции зон внедрения и разветвления нервных волокон в мышцу) были идентифицированы и отмечены опытным физиотерапевтом.
За 48 часов и за 24 часа до введения МСККМ выполнена подкожная инъекция гранулоцитарного колониестимулирующего фактора (Г-КСФ, 300 мкг). Выполнена трансплантация собственных МСККМ пациента в положении лежа. Костный мозг был получен из передней верхней части правой подвздошной кости. Изолированные мононуклеарные клетки периферической крови (МКПК) были проверены на наличие CD34+ маркеров с помощью метода проточной цитометрии, разведены в спинномозговой жидкости и введены внутримышечно в двигательные точки и в полость позвоночного канала. Для уменьшения воспаления внутривенно вводился метилпреднизолон (1мг) разведенный в растворе Рингера (500 мл). После клеточной терапии пациент прошел курс нейрореабилитации. Мультидисциплинарной протокол включал в себя физиотерапию, трудотерапию, консультацию психолога и советы по питанию. Физиотерапия включала в себя упражнения для укрепления слабых мышц, силовые упражнения для неповрежденных мышц, коррекцию походки, поддержание равновесия. Трудотерапия включала в себя функциональную тренировку, реабилитацию рук и их шинирование для предотвращение деформации. Пациенту была рекомендована регулярная терапия на дому и диета с высоким содержанием белков.
Результаты
После клеточной терапии пациент дважды наблюдался в стационаре через 3 и 9 месяцев. Спустя 3 месяца было отмечено улучшение двигательной функции верхних конечностей. Выполнение движений над головой требовало сравнительно меньших усилий. Наблюдалось двустороннее снижение жесткости и псевдогипертрофии икроножных мышц . Отмечались значительные улучшения в положении стоя и сидя, в способности держать равновесие. Баланс в положении стоя и при ходьбе улучшился. Частота падений при ходьбе заметно уменьшилась с 4-5 падений в месяц до 1 падения за 3 месяца. Характеристики дыхательной функции также улучшились: жизненная емкость легких (ЖЕЛ) (с 1250 мл до 1750 мл) и пиковая скорость выдоха (ПСВ) (с 290 мл до 360 мл),.
Спустя 9 месяцев пациент также находился под наблюдением врачей и никаких ухудшений по описанным выше симптомам не было. Его выносливость во время выполнения упражнений и в повседневной активности увеличилась. Вместе с тем снизились и уровни утомляемости и появилась возможность выполнять упражнения с большей легкостью. Модифицированная шкала ручного тестирования мышц Совета по медицинским исследованиям (далее также – mMRC-MMT) показала значительное улучшение почти во всех группах мышц (Таблица 2).
| Группы мышц | До введения стволовых клеток | После введения (спустя 3 месяца) | После введения (спустя 9 месяцев) | |
| Бедра | Сгибатели | 3- | 3+ | 3+ |
| Разгибатели | 2- | 2- | 2 | |
| Отводящие мышцы | 2+ | 2+ | 2++ | |
| Приводящие мышцы | 2- | 2+ | 2+ | |
| Колено | Сгибатели | 2++ | 2++ | 2++ |
| Разгибатели | 2++ | 2++ | 2++ | |
| Лодыжки и стопы | Большеберцовая мышца | 3+ | 3++ | 3++ |
| (Задняя голень) | 4 | 4 | 4 | |
| Подошвенные сгибатели | 4 | 4 | 4 | |
| Длинный разгибатель пальцев | 3- | 3 | 3 | |
| Туловище | Верхняя часть живота | 2+ | 2+ | 2+ |
| Нижняя часть живота | 2++ | 3+ | 3+ | |
| Разгибатели спины | 1+ | 2 | 2 | |
| Шея | Трапециевидная мышца | 4 | 4 | 4 |
| Ромбовидные мышцы | 3++ | 3++ | 3++ | |
| Передняя зубчатая мышца | 3+ | 3+ | 3+ | |
| Плечи | Сгибатели | 3++ | 3++ | 3++ |
| Разибатели | 3+ | 3++ | 3++ | |
| Отводящие мышцы | 3++ | 3++ | 3++ | |
| Приводящие мышцы | 3++ | 3++ | 3++ | |
| Наружные ротаторы | 3+ | 3++ | 3++ | |
| Внутренние ротаторы | 3+ | 3++ | 3+ | |
| Руки | Бицепс | 3+ | 3++ | 3++ |
| Плечо | 3+ | 3++ | 3+ | |
| Трицепс | 3+ | 3++ | 3++ | |
| Плечелучевая | 3+ | 3++ | 3++ | |
| Предплечье запястье, рука | ||||
Шкала “Северной Звезды” показала значительное улучшение во время второго наблюдения. Остальные показатели шкал, упомянутые выше, после вмешательства остались неизменными, что может означать прекращение прогрессии заболевания.
О мышечной дистрофии Беккера и клеточной терапии
МДБ медленно приводит к инвалидности вследствие снижения подвижности и способности самостоятельного ухода. Это может выражаться в сердечных проявлениях, таких как: учащенное сердцебиение, головокружение, обморок, одышка в состоянии покоя или во время физической нагрузки.
Ведение МДБ мультидисциплинарное и состоит из медицинского ухода с использования кортикостероидов, которые снижают воспалительный процесс мышечных волокон.
Реабилитация подразумевает продление периода функциональной самостоятельности. Она направлена на симптомы, а не на причину заболевания. В настоящее время нет определенной терапевтической стратегии для контроля прогрессии заболевания и увеличения мышечной силы.
Генная терапия нацелена на введение отсутствующего гена дистрофина, используя различные векторы. Ряд практических трудностей не позволяет генной терапии быть клинически применимым методом в настоящее время. Трансплантация стволовых клеток предлагается в качестве лечения таких заболеваний, как мышечная дистрофия. На основе клеточной терапии предпринимались попытки стимулировать регенерацию мышц с надеждой на то, что стволовые клетки восстановят мышечную функцию и исправят патологию путем повторного синтеза мышц. Стволовые клетки рассматриваются как подходящий вариант для терапевтического применения из-за их способности к самовосстановлению и потенциала дифференцировки. В последние годы были получены обнадеживающие результаты лечения человека с использованием зрелых стволовых клеток.
Alok Sharma при участии соавторов., в 2013 году изучали эффект внутримышечной аутотрансплантации мезенхимальных стволовых клеток костного мозга у 150 пациентов с мышечной дистрофией. Через 12 месяцев наблюдения у пациентов наблюдалось увеличение мышечной силы и улучшение походки. Симптоматические и функциональные улучшения также наблюдались в 86,67% случаев: у шести пациентов снижен уровень жировой инфильтрации и выявлена регенерация мышц на МРТ , а у девяти – выявлены положительные изменения электрической активности мышц на ЭМГ.
Мезанхимальные стволовые клетки состоят из множества клеток, таких как гематопоэтические стволовые клетки, тканеспецифические клетки-предшественники, стромальные клетки и специализированные клетки крови на разных стадиях развития. Эти клетки обладают способностью мобилизовать и оказывать свои репаративные эффекты в месте повреждения. Они способствуют неоваскуляризации и усиливают ангиогенез (образование кровеносных сосудов), продуцируя сигнальные молекулы, такие как факторы роста эндотелия сосудов и факторы роста фибробластов (FGF2). Они также способствуют ремоделированию тканей, предотвращают апоптоз (отмирание клеток), уменьшают воспаление, высвобождают факторы роста и активируют сателлитные клетки. Это паракринные эффекты, которые могут помочь в достижении желаемого результата клеточной терапии. Аутологичные мезенхимальные стволовые клетки костного мозга были использованы в этом случае, потому что они не имеют этических проблем, и его безопасность была установлена (не требуется донор, это клетки самого пациента).
Трансплантация стволовых клеток в нужное место мышечного тела, как правило, является основной практической трудностью. Внутривенное введение стволовых клеток, полученных из костного мозга, показало успешное возвращение стволовых клеток в поврежденные мышечные ткани на моделях животных; однако это созадет риск разбавления концентрации клеток. Мышечная дистрофия в основном воспринимается как заболевание мышц, с малым количеством свидетельств нервно-мышечных поражений. Дистрофин является частью структурного белка, обнаруженного в миелине, образующего клетки Шванна и нервы. Демиелинизация и дегенерация как изменения в нервах могут происходить с такими нарушениями в клетках. Поэтому были выбраны два различных способа трансплантации клеток: внутримышечный и интратекальный. Мезенхимальные клетки костного мозга вводили в двигательные точки целевых слабых мышц для восстановления иннервирующего нерва, а также мышц. Известно, что спинномозговая жидкость содержит факторы роста, которые помогают росту коркового эпителия и стимулируют васкуляризацию в нервной системе, поэтому она использовалась в качестве разбавляющей среды.
Мышечная сила регистрировалась с помощью ручного тестирования мышц по шкале, разработанной опытными физиотерапевтами исследовательской группы на основе модифицированной шкалы ручного тестирования мышц Совета по медицинским исследованиям (mMRC-MMT). Поскольку mMRC-MMT не подразделяется на 1 и 2 классы на основе частичного диапазона движения (ЧДД), в данном случае в шкалу mMRC MMT – I добавлены 1 и 2 классы. Это позволило исследовательской группе количественно оценить минимальные изменения силы, наблюдаемые у пациентов с МДБ (Таблица 2). Увеличение мышечной силы, которое было зарегистрировано в mMRC-MMT, также наблюдалось в элементах шкалы “Северной звезды”, таких как подъем и сидение. Функционально и баланс в положении стоя и при ходьбе были улучшены. Все объективные показатели не показали ухудшений.
Важной причиной заболеваемости и смертности при мышечной дистрофии может быть нарушение функции дыхания, но в данном исследовании было отмечено улучшение значений максимального объема вдоха и PEFR по сравнению с исходным уровнем, а также было снижение уровня усталости и улучшение выносливости во время действий, которые могут быть связаны с улучшением функции дыхательных мышц.
Для поддержания достигнутых улучшений и для статического прогрессирования может быть полезным повторение процедуры клеточной терапии. Клеточная трансплантация может вызвать регенерацию дегенерированных мышц и может изменить прогрессирование заболевания при МДБ. Несмотря на то, что данный клинический случай является результатом наблюдения за одним пациентом, он может послужить важным шагом в проведении дальнейших исследований. Для установления оптимальной дозировки, источника и частоты трансплантации необходимы тщательный анализ и крупные клинические испытания со сложной методологией. Одно из ограничений исследования заключается в том, что у него нет похожего клинического случая для сравнения, но поскольку у пациента наблюдалась остановка прогрессии после клеточной терапии, можно предположить, что клеточная трансплантация сыграла в этом жизненно важную роль.
В заключение, клеточная терапия в сочетании с реабилитацией может предложить возможность восстановления и регенерации мышечных волокон, снижая при этом скорость прогрессирования Мышечной дистрофии Беккера.
Перевод выполнен: Бережной Д.С., Чернец Е.Н.
Читайте также:
