
Мышечная дистрофия дюшенна википедия это генетическая болезнь
Существует огромное количество различных заболеваний, которые возникают у деток независимо от обстоятельств или действия окружающей среды. Это категория именно наследственных болезней. Сейчас же пойдет речь о такой проблеме, как мышечная дистрофия Дюшенна: что это за хворь такая, каковы у нее симптомы и можно ли с ней справиться.
Терминология
Изначально нужно узнать, что же такое наследственные болезни. Так, это заболевания, которые возникают в результате дефектов аппарата наследственных клеток. То есть это определенные сбои, которые происходят на генетическом уровне.
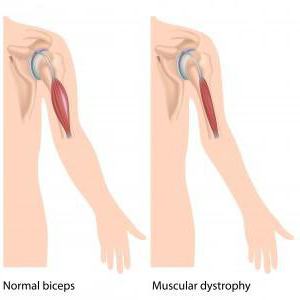
Мышечная дистрофия Дюшенна – это именно наследственная болезнь. Проявляется она очень быстро, основной симптом в данном случае – это быстро прогрессирующая слабость в мышцах. Нужно отметить: как и все остальные мышечные заболевания, болезнь Дюшенна также приводит в конечном результате к атрофии мышц, нарушению моторики и, конечно же, инвалидности. В подростковом возрасте детки с таким диагнозом уже не имеют возможности самостоятельно передвигаться и не могут обходиться без посторонней помощи.
Что происходит на генном уровне
Как уже было отмечено, мышечная дистрофия Дюшенна – это генетическое заболевание. Так, происходит мутация в том гене, который отвечает за выработку особого белка дистрофина. Именно он и необходим для нормальной работы мышечных волокон. При этом важно отметить, что эта генетическая мутация может как передаваться по наследству, так и возникать спонтанно.
Также важно отметить, что ген локализируется в хромосоме Х. Но женщины этой болезнью заболеть не могут, являясь только лишь передатчиком мутации от поколения к поколению. То есть если мама передаст мутацию сыну, он с 50%-й вероятностью заболеет. Если же девочке, она просто будет носителем гена, клинических проявлений болезни у нее не будет.
Симптоматика: группы
В основном, болезнь активно заявляет о себе примерно в 5-6 летнем возрасте. Однако первые симптомы могут возникнуть у малыша, который еще не достиг трехлетнего возраста. При этом надо отметить, что все патологические нарушения медки условно разделяют на несколько больших групп:
- Поражение мускулатуры.
- Поражение сердечной мышцы.
- Деформация скелета ребенка.
- Различные эндокринные расстройства.
- Нарушения нормальной умственной деятельности.
Наиболее часто встречающиеся проявления болезни
Обязательно также надо рассказать о том, как проявляется синдром Дюшенна. Симптомы бывают следующие:
- Слабость. Которая постепенно нарастает, развивается.
- Начинается прогрессирующая мышечная слабость именно с верхних конечностей, далее затрагиваются ноги и только потом – все остальные части тела и органы.
- Ребенок утрачивает возможность сам передвигаться. Примерно к 12-летнему возрасту такие детки уже полностью зависимы от инвалидной коляски.
- Также наблюдаются расстройства дыхательной системы.
- Ну и, конечно же, бывают нарушения в работе кардиологической системы. Позже происходят необратимые изменения в миокарде.
О поражении мышц скелета
Именно поражения мышечной ткани – наиболее распространенный симптом, если речь идет о такой проблеме, как синдром Дюшенна. При этом надо отметить, что рождаются детки без особых отклонений в развитии. В малом возрасте ребята менее активны и подвижны, нежели сверстники. Но чаще всего это связывают с темпераментом и характером ребенка. Поэтому отклонения очень редко замечаются. Более существенные признаки проявляются уже во время ходьбы малыша. Такие детки могут передвигаться на носочках, не становясь на полную стопу. Также они частенько падают.
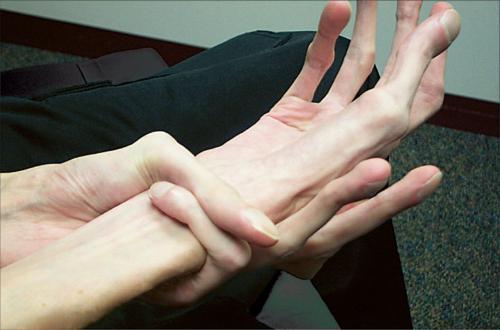
Особым показателем является еще и симптом Говерса. То есть ребенок, чтобы подняться с пола, активно пользуется руками, как бы взбираясь по самому себе.
Также надо отметить, что при такой проблеме, как синдром Дюшенна, у ребенка постепенно атрофируются мышцы. Но нередко бывает так, что у крохи внешне мускулатура кажется очень развитой. Мальчик даже на первую вскидку оказывается как бы накачанным. Но это только лишь обман зрения. Все дело в том, что в процессе болезни мышечные волокна постепенно распадаются, а их место занимает жировая ткань. Отсюда и такой внушительный внешний вид.

Немного о деформации скелета
Если у ребенка прогрессирующая мышечная дистрофия Дюшенна, то постепенно у мальчика изменится форма скелета. Сначала патология затронет поясничный отдел, далее возникнет сколиоз, то есть произойдет искривление грудного отдела позвоночника. Позже проявится сутулость и, конечно же, изменится нормальная форма стопы. Вся данная симптоматика еще в большей степени будет сопутствовать ухудшению двигательной активности малыша.
О сердечной мышце
Обязательным симптомом при данной болезни также является поражение сердечной мышцы. Происходит нарушение ритма сердца, возникают регулярные перепады артериального давления. При этом сердце увеличивается в размерах. Но его функциональные возможности наоборот, уменьшаются. И в результате постепенно формируется сердечная недостаточность. Если эта проблема еще будет сочетаться с дыхательной недостаточностью, то возникает большая вероятность летального исхода.

Нарушения умственной активности
Нужно отметить, что мышечная дистрофия Дюшенна-Беккера не всегда проявляется таким симптомом, как умственная отсталость. Связано это может быть с дефицитом такого вещества, как аподистрофин, необходимого для работы головного мозга. Нарушения интеллекта могут быть самыми разными – от слабой умственной отсталости до идиотии. Усугублению этих когнитивных расстройств способствует еще и невозможность посещать садики, школы, кружки и иные места скопления детей. В результате возникает социальная дезадаптация.
Расстройства работы эндокринной системы
Различные эндокринные расстройства встречаются не более чем у 30-50% всех больных. Чаще всего это именно лишний вес, ожирение. При этом детки также имеют более низкий, чем у сверстников, рост.
Исход болезни
Какова клинико-эпидемиологическая характеристика мышечной дистрофии Дюшенна? Так, частота возникновения болезни – 3,3 пациента на 100 тысяч здоровых людей. Нужно отметить, что мышечная атрофия постепенно прогрессирует, и к 15-летнему возрасту мальчик уже не может обходиться без помощи окружающих, являясь полностью обездвиженным. Ко всему, происходит еще и частое присоединение различных бактериальных инфекций (чаще всего именно мочеполовой и дыхательной систем), при неправильном уходе за ребенком возникают пролежни. Если проблемы с дыхательной системой соединяются с сердечной недостаточностью, это грозит смертельным исходом. Если же говорить в общем, то такие пациенты практически никогда не живут более 30 лет.
Диагностика болезни
- Генетическое тестирование, то есть анализ ДНК.
- Электромиография, когда подтверждается первичное изменение мышц.
- Биопсия мышц, когда происходит определение наличия белка дистрофина в мышце.
- Анализ крови на определение уровня креатинкиназы. Нужно отметить, что именно этот фермент указывает на гибель мышечных волокон.
Лечение
Полностью излечиться от данной болезни невозможно. Можно только облегчить проявление симптомов, что сделает жизнь больного немного проще и удобнее. Так, после того, как пациенту ставят такой диагноз, чаще всего ему назначают терапию глюкокортикостеродами, которые призваны замедлить процесс развития болезни. Иные процедуры, которые также могут быть использованы при данной проблеме:
- Дополнительная вентиляция легких.
- Терапия медикаментами, которая направлена на нормализацию работы сердечной мышцы.
- Использование различных приспособлений, которые повышают мобильность пациента.
Также важно отметить, что сегодня ведутся разработки новейших методик, которые основаны на генной терапии, а также пересадке стволовых клеток.

Иные мышечные заболевания
Существуют еще и иные мышечные врожденные заболевания детей. К таким болезням можно отнести, помимо дистрофии Дюшенна:
- Дистрофию Беккера. Эта болезнь очень похожа на синдром Дюшенна.
- Мышечную дистрофию Дрейфуса. Это медленно прогрессирующая болезнь, при которой интеллект сохраняется.
- Прогрессирующую мышечную дистрофию Эрба-Рота. Проявляется в подростковом возрасте, прогрессирование быстрое, инвалидизация наступает рано.
- Плечелопаточно-лицевую форму Ландузи-Дежерина, когда мышечная слабость локализируется в области лица, плеч.
При этом надо отметить, что ни при одной из этих болезней не проявляется мышечная слабость у новорожденных. Вся симптоматика возникает в основном в подростковом возрасте. Длительность жизни пациентов чаще всего не превышает 30 лет.

Мышечная дистрофия, или миопатия Дюшенна, — тяжелая наследственная патология, которая постоянно прогрессирует. Замедлить мышечное разрушение практически невозможно.
Связано это с врожденными изменениями. Впервые о миопатии Дюшенна заговорили в середине XIX века. Обнаружил эту патологию французский невролог. В тот момент был известен один тип течения болезни, через некоторое время выделили еще несколько способов развития состояния.
Этот тип болезни сильно похож на миодистрофию Беккера, но в то же время отличается от него сложностью и внешними признаками.
Миодистрофия Дюшенна обнаруживает у 1 ребенка из 4000. Этот тип патологии относится к самым распространенным мышечным дистрофиям, относится к врожденным заболеваниям.
Этиология нарушения
Одному из генов в структуре генома человека присвоили имя невролога, в честь которого и было названо отклонение. На мышечную дистрофию Дюшенна могут влиять разные факторы:
- кровосмешение;
- предрасположенность генетического характера, например, при наличии миопатии Дюшенна у кого-либо из родни;
- неправильный синтез мышечных волокон, ускоренное распространение и замещение жировой прослойкой, соединительными волокнами;
- наследственные формы синдрома Дюшенна, чаще всего переходящие от матери;
- мутация генома при формировании во время беременности;
- аномалии в хромосомных структурах неясного происхождения;
- сильные нарушения в развитии дистрофина;
- патологические изменения биохимии в крови.

Характеристика наследственной патологии
Генетическая природа заболевания была сразу же доказана после обнаружения синдрома в 1868 году. Эта патология почти идентична с миодистрофией Беккера, то есть, обладает теми же генетическими предпосылками для формирования.
Однако миодистрофия Беккера отличается иными симптомами. Для болезни характерны следующие особенности:
- диагностируется у мальчиков до 5 лет;
- прогрессирует стремительно;
- у девочек никогда не обнаруживается;
- атрофия мышц обладает ступенчатым развитием – сначала страдает тазовый пояс;
- затем вовлекаются мышцы ног;
- после этого миопатия Дюшенна поражает мышцы спины, плеч;
- завершается прогрессирующая мышечная дистрофия Дюшенна поражением рук;
- специфический признак нарушения – деформация позвоночника, чаще встречающаяся в форме кифоза или лордоза;
- миодистрофия Дюшенна почти всегда сопровождается повреждениями грудины и стоп, они становятся неправильной формы, сильно меняют тело человека;
- при патологии, в отличие от миодистрофии Беккера, появляется повреждение левого сердечного желудочка, аритмия и кардиопатия;
- примерно у 30% пациентов развивается олигофрения.

Мышечная дистрофия Дюшенна никогда не протекает в легкой степени, всегда имеет крайне неблагоприятный прогноз. Развивается быстро, возможность ходить пациент утрачивает уже к 12 годам. При мышечной дистрофии Дюшенна смерть наступает из-за инфекции бронхов или легких, после остановки сердца.
Симптомы нарушения
Первые признаки миопатии Дюшенна встречаются уже в возрасте 1,5 лет. В редких случаях их не удается заметить до 5 лет. Проявляются признаки заболевания Дюшенна сначала в легкой степени. Их комбинация зависит от общего состояния здоровья:

На фоне мышечной дистрофии Дюшенна у маленького пациента развивается острая депрессия, которую дети с трудом переносят. Нередко причиной смертности при миодистрофии Беккера и Дюшенна становится суицид.
Диагностика заболевания
Мышечная дистрофия Дюшенна крайне тяжело поддается диагностики. Для этого привлекают комплекс методов. Первое, что нужно пройти при подозрении на миопатию Дюшенна, — это ЭКГ. Для подтверждения диагноза необходимо, чтобы анализ показал нарушения стенки левого желудочка.

Следующий этап – это определение уровня дистрофина, который не меняется в сторону обычной дистрофии. Также необходимо сдать кровь на биохимический анализ. Если есть миодистрофия Беккера или болезнь, названная в честь французского невролога, отмечается высокий уровень КФК.

Тактика лечения заболевания
Чтобы лечение мышечной дистрофии Дюшенна было эффективным, нужно четко следовать намеченному плану после постановки диагноза. Излечению болезнь никогда не поддается полностью, но можно значительно облегчить жизнь пациента. Современная медицина способна замедлить миопатию Дюшенна следующими методами:

Важно! Поддержать здоровье при мышечной дистрофии Дюшенна можно и другими методами – ЛФК и электрофорезом.

В тяжелых случаях всё лечение проводят в домашних условиях, если есть медицинские возможности для организации сложной терапии специальными приборами.
Обязательное условие для лечения миопатии Дюшенна – постоянное наблюдение у кардиолога. Также необходимо составить грамотное меню. При заболевании нужно есть много овощей, приготовленных на пару, фруктов, растительных жиров и нежирного мяса. Запрещено употребление алкоголя, кофеина и крепкого чая.
Последствия и осложнения
В 100% случаев миопатия Дюшенна сопровождается тяжелыми последствиями для организма и сильно укорачивает жизнь. Пациент всегда умирает от осложнений заболевания – остановки сердца или легочной инфекции.
Если мышечную дистрофию Дюшенна удалось обнаружить в раннем возрасте, есть шанс, что человек доживет до 30 лет. Но только при условии адекватной терапии и комплексного подхода. Среди осложнений миопатии Дюшена нередко выделяют остеопороз, поражения позвоночника и суставов, а также патологии пищеварительной системы.
Это заболевание поражает примерно 1 человека из 4000, то есть - это наиболее распространенный тип мышечной дистрофии. Обычно на МДД болеют только мужчины, хотя женщины могут иногда быть носителями заболевания. Если же отец болен МДД, а мать является носителем, или тоже больна, то в таком случае на мышечную дистрофию Дюшенна может заболеть женщина. Расстройство возникает в связи с мутацией в гене дистрофин, который у людей расположен на Х-хромосоме (Xp21). Ген дистрофин кодирует деятельность белка дистрофина, который является важной структурной составляющей мышечной ткани. Дистрофин обеспечивает структурную устойчивость дистрофин-ассоциированного-гликопротеинового комплекса (ДАГ комплекса), расположенного на клеточной мембране.
Симптомы заболевания обычно появляются у детей мужского пола до 5 лет и могут проявиться в раннем детстве. Первыми признаками болезни являются прогрессирующая проксимальная слабость мышц ног и таза, связанная с потерей мышечной массы. Постепенно эта слабость распространяется на руки, шею и другие части тела. Ранние признаки расстройства могут также включать псевдогипертрофию (увеличение икроножных мышц и дельтовидных мышц), низкую выносливость и трудности при стоянии без посторонней помощи, как правило, человек также не может самостоятельно подняться. В процессе прогрессирования заболевания мышечная ткань постепенно заменяется жировой и фиброзной тканями (как следствие развивается фиброз). Для помощи при ходьбе, в возрасте 10 лет может быть необходимым применение специальных подтяжек, но большинство пациентов уже старше 12 лет не могут передвигаться без инвалидной коляски.
Позже возникают следующие признаки расстройства: аномалии развития костей, которые приводят к деформации скелета, в том числе к искривлению позвоночника.
В связи с прогрессивным ухудшением работы мышц, особь теряет возможность двигаться, что в конечном счете, может быть причиной паралича. Относительно отклонений в психическом развитии, то их наличие зависит от каждого конкретного случая, но если определенные отклонения и присутствуют, то они несущественно влияют на развитие ребенка, ведь со временем нарушение не прогрессирует. Средняя продолжительность жизни больных МДД варьирует от подросткового возраста до 20 - 30 лет. Известны случаи, когда больные доживали до 40 лет, но, к сожалению, такие случаи являются скорее исключением.
Распространенность
Мышечная дистрофия Дюшенна возникает в связи с мутациями в гене дистрофин, который расположен на Х-хромосоме. В связи с этим, ДМД встречается у 1 человека на 4000 новорожденных мужского пола. Мутации в гене дистрофин могут быть унаследованы или возникают спонтанно во время зародышевой линии передачи.
Эпоним
Заболевание названо в честь французского невропатолога Жульема Бенджамина Аманда Дюшенна (Guillaume Benjamin Amand Duchenne), который впервые описал это заболевание в 1861 году.
Патогенез
Мышечная дистрофия Дюшенна обусловлена мутацией в гене дистрофин, локус которого Xp21. Дистрофин отвечает за соединение цитоскелета каждого мышечного волокна с основной базальной пластинкой (внеклеточного матрикса) через белковый комплекс, который состоит из многих субъединиц. Отсутствие дистрофина приводит к проникновению избыточного кальция в сарколему (клеточную мембрану). Как следствие изменения этих сигнальных путей, вода наполняет митохондрии, которые после этого разрываются. При дистрофии скелетных мышц, митохондриальная дисфункция приводит к усилению стресса вызванного цитозольным-кальциевым сигналом и усилению производства стресс-индуцированных активных форм кислорода (АФК). В этом сложном каскадном комплексе, который включает в себя несколько реакций еще до сих пор не понятно до конца, почему из-за повреждения сарколеммы увеличиваются проявления окислительного стресса, который в итоге приводит к смерти клетки. Мышечные волокна подвергаются некрозу и, наконец, происходит замена мышечной ткани жировой, а также соединительной.
Симптомы
Основным симптомом мышечной дистрофии Дюшенна - является мышечная слабость, которая в первую очередь связана с атрофией мышц, а именно скелетной мышечной ткани. В первую очередь атрофируются мышцы бедер, таза, плеч и икроножные мышцы. Мышечная слабость возникает также в руках, шее и других частях тела, но обычно не так рано, как в нижней части тела. Икры часто увеличены. Обычно симптомы появляются в возрасте до 6 лет, но могут впервые проявиться еще в раннем детстве.
Признаки и тестирования
Как уже было сказано, атрофия мышц при МДД начинается в виде мышечной слабости в ногах и тазовом поясе, затем переходит к мышцам плеч и шеи, после чего, повреждает мышцы рук и дыхательные мышцы. Важным видимым признаком в начале развития заболевания является увеличение икроножных мышц (псевдогипертрофии). Распространенным явлением есть кардиомиопатия, но развитие сердечной недостаточности или аритмии (заболевания связаны с нарушениями ритма сердца, последовательности и силы сокращений сердечной мышцы) встречаются довольно редко.
- наличие симптома Говерса отражает более тяжелые нарушения мышц нижних конечностей. Можно говорить о наличии симптомов, в случае, если ребенок помогает себе встать с помощью рук: сначала, ребенок становится на четвереньки (опираясь на пол ногами и руками), а затем, держась руками за ноги, контролирует направление своего движения;
- больные МДД дети, часто устают быстрее и имеют меньше силы, чем их сверстники;
- очень высокий уровень креатин-киназы (КФК-ММ) в крови тоже может стать показателем развития и прогрессирования заболевания;
- при проведении электромиографии (ЭМГ) видно, что слабость организма вызвана повреждением мышечной ткани, а не повреждением нервной проводимости;
- генетическое тестирование может выявить генетические нарушения в гене Xp21;
- мышечная биопсия с последующим гистологическим, иммуногистохимическим или имуноблотинговим исследованием) или генетическое тестирование (с помощью анализа крови), подтверждает отсутствие дистрофин.
ДНК-тест
Мышечно-специфическая изоформа гена дистрофина состоит из 79 экзонов. Тестирование ДНК и их анализ, как правило, позволяют определить тип мутации экзона или определить какие экзоны повреждены. Анализ ДНК в большинстве случаев подтверждает предварительную диагностику другими методами.
Биопсия мышц
Если при анализе ДНК никаких мутаций не обнаруживается, то возможно проведение мышечной биопсии. Для этой процедуры с помощью специального инструмента берут маленький образец мышечной ткани и, использовав специальный краситель, определяют наличие /отсутствие в мышечной ткани дистрофина. Полное отсутствие белка указывает на наличие этого заболевания.
За последние несколько лет ДНК-тесты были существенно усовершенствованы, на сегодня они проявляют больше мутаций и поэтому мышечную биопсию для подтверждения МДД сейчас используют все реже.
Пренатальное тестирование
Если один или оба родителя являются "носителями" этого заболевания, то существует риск того, что их еще не народившийся ребенок будет поражен этим расстройством. Для определения того, будет будущий ребенок больным МДД, используют методы пренатальной диагностики. На сегодня эти методы доступны только для определения некоторых нервно-мышечных расстройств. Различные пренатальные тесты могут проводиться примерно на 11 недели беременности.
Исследование с помощью биопсии хориона (CVS) можно проводить на 11-14 неделях, амниоцентез можно использовать после 15 недели, забор крови плода возможен примерно на 18 неделе. Родители должны внимательно изучить все возможные методы и, возможно, с помощью генетического консультанта выбрать наиболее оптимальный для себя вариант. Если тестирование будет осуществлено на ранних сроках беременности, то это позволит досрочно прекратить беременность, в случае наличия заболевания у плода, однако, при применении таких методов, увеличивается риск выкидыша при последующих беременностях, чем при тех методах, которые применяются позже (около 2% , по сравнению с 0,5%).
Лечение
Никаких известных эффективных препаратов для лечения мышечной дистрофии Дюшенна, не существует. Хотя согласно последним исследованиям стволовых клеток существуют перспективные векторы, которые могут заменить поврежденные мышечные ткани. Однако, на данном этапе лечения, как правило, симптоматическое и направлено на улучшение качества жизни больного человека.
Оно включает в себя:
- употребление таких кортикостероидов как преднизолон и дефлазакорт для увеличения энергии и сил и облегчения тяжести некоторых симптомов;
- рандомизированные контролируемые исследования показывают, что использование бета 2-агонистов увеличивает мышечную силу, но не замедляет процесс прогрессирования заболевания. Время контроля за людьми, которые употребляли бета 2-агонисты составляет около 12 месяцев, следовательно, результаты этих испытаний не могут быть экстраполированы на больший период времени;
- рекомендуется умеренная физическая активность, разрешается заниматься плаванием. Бездействие (например, постельный режим) может усилить прогрессирование заболевания;
- для поддержания мышечной силы, гибкости и функциональности суставов важна физиотерапия;
- использование ортопедических приспособлений (например, инвалидных колясок) может улучшить способность больного двигаться и самостоятельно обеспечивать свои потребности. Использование так называемых съемных стяжек, фиксирующих голень во время сна позволяет отложить начало контрактур (ограничение движений суставов).
- по мере прогрессирования заболевания необходимым становится использование специальных респираторных механизмов, позволяющих обеспечить нормальный процесс дыхания.
Центром по контролю и профилактике заболеваний (Centers for Disease Control and Prevention (CDC)) были разработаны общие многопрофильные стандарты (принципы) помощи больным МДД. Эти принципы были опубликованы в двух частях в журнале The Lancet Neurology в 2010 году. (Http://www.treat-nmd.eu/patients/DMD/dmd-care).
Прогноз
Мышечная дистрофия Дюшенна повреждает все скелетные мышцы, мышцы сердца и дыхательные мышцы (на более поздних стадиях). Больные МДД, как правило, живут только к подростковому возрасту или умирают в возрасте 30-40 лет. Последние достижения в области медицины, позволяют надеяться на увеличение продолжительности жизни больных этим расстройством.
Иногда (но очень редко) особи с МДД доживали до 40-50 лет, но лишь с помощью использования надлежащего дополнительного оборудования (инвалидных колясок и кроваток), вентиляционной поддержки дыхания (с помощью трахеостомии или специальной дыхательной трубки), очистки дыхательных путей и принятие необходимых сердечных препаратов. Кроме того, для увеличения продолжительности жизни необходимо на ранних этапах заболевания спланировать механизм ухода за больным на более поздних этапах.

Миопатия Дюшенна — наследственная патология, характеризующаяся прогрессирующим нарушением клеточного метаболизма в мышечной ткани, которое приводит к ее структурным изменениям и разрушению. Заболевание развивается только у мальчиков. Женщины – носители мутантного гена. Симптоматика болезни нарастает постепенно и начинает проявляться с самого детства. Родители замечают, что ребенок плохо стоит, с трудом карабкается на лесенку, часто падает во время ходьбы.
Миопатия была впервые описана в середине 19 века неврологом из Франции Г. Дюшенном. Он лечил больных с признаками мышечной патологии, анализировал наблюдения и представил свои умозаключения научному сообществу. Врач доказал генетическую природу заболевания.

Мышечная слабость прогрессирует постепенно. Симптомами заболевания являются: специфическая походка, неправильная осанка, позднее физическое и психическое развитие, утрата приобретенных двигательных навыков. У детей возникают проблемы с ходьбой и бегом. С возрастом признаки патологии становятся более выраженными и разнообразными. Симптомы усугубляются под воздействием негативных факторов – психофизического перенапряжения, острых инфекций, отравления. Они резко снижают качество жизни лиц с миопатией. У больных отмечается гипо- и арефлексия, искривление позвоночника, килевидная или седловидная грудная клетка, контрактуры суставов и ретракции сухожилий. Часто к поражению опорно-двигательного аппарата присоединяются проблемы с сердцем в виде кардиомиопатии и изменения интеллекта в виде олигофрении и дебильности.

Диагностика миодистрофии Дюшенна заключается в проведении медико-генетического исследования, которое позволяет выявить мутантный ген. Больным проводят биопсию мышечной ткани для определения в ней уровня белка дистрофина. Данное наследственное заболевание неизлечимо. Побороть недуг невозможно. Специалисты проводят симптоматическую и паллиативную терапию, облегчающую и продлевающую жизнь больным. Медикаментозное лечение глюкокортикостероидами, ЛФК, массаж и физиопроцедуры в комплексе дают относительно неплохие результаты.
Заболевание отличается тяжелым, злокачественным течением и неблагоприятным прогнозом. Больные рано перестают самостоятельно двигаться, с 8-10 лет используют костыли, а с 12 – инвалидные коляски. Шестнадцатилетние подростки испытывают нарушения со стороны органов дыхания и сердца, у некоторых снижается интеллект. Нарастающая мышечная слабость приводит к полной обездвиженности больного. Замедлить разрушение мышц невозможно, что связано с врожденным характером патологических изменений. Лица с миодистрофией умирают в возрасте 25-30 лет от инфекционной пневмонии или острой коронарной недостаточности.
Этиология

Миопатия Дюшена — генетически детерминированный недуг, наследуемый по рецессивному принципу, сцепленному с Х-хромосомой. Слабость мышц развивается у мальчиков. Девочки являются носителями мутантного гена. Именно они в 70% случаев передают патологию своим сыновьям. В оставшихся 30% мутации возникают спонтанно во время внутриутробного развития плода. Причины подобных мутаций неизвестны. Чаще всего это случается в близкородственных браках или у лиц, имеющих иные генетические аномалии.
Мутантный ген, обуславливающий развитие миопатии, получил название ген Дюшенна. Он отвечает за синтез белка дистрофина, который обеспечивает целостность мышечных волокон во время сокращения. Белок составляет основу миофибрилл, поддерживает их клеточный скелет, позволяет им активно и многократно сокращаться и расслабляться. У лиц с миопатией он отсутствует или вырабатывается в очень незначительном количестве. Мышечные волокна постепенно разрушаются и замещаются фиброзной тканью или жиром. В результате серьезно страдает функция движения у больных.
Симптоматика
Новорожденные дети не имеют каких-либо явных отклонений в здоровье и строении. Первые клинические признаки появляются у малышей 1,5-2 лет. Болезнь прогрессирует, а симптоматика нарастает.

проявления миопатии Дюшенна
- Родители замечают, что их ребенок двигается неловко и неустойчиво, постоянно спотыкается и падает при ходьбе, не может прыгнуть, подняться по ступенькам, встать из лежачего или сидячего положения. Больные дети очень медлительны и неуклюжи во время двигательной активности. Это связано с поражением мышц таза и ягодиц, которые становятся слабыми первыми.
- Появляется характерный симптом Говерса: больные, поднимаясь с пола, помогают себе руками – они опираются на свои колени и бедра.

Больные дети рано становятся инвалидами, прикованными к креслу. К 10-12 годам они полностью утрачивают способность к самостоятельному передвижению, а к 15 — к совершению любых действий. В течение пяти лет после этого атрофируются дыхательные мышцы.
Погибают больные обычно в возрасте 25 лет от неуклонно нарастающей слабости скелетных мышц, сформировавшейся стойкой дисфункции органов дыхания и сердечно-сосудистой системы.
Осложнения
Мышечная дистрофия Дюшенна сокращает жизненный путь человека. Это основное и самое страшное последствие болезни.
Осложнения со стороны опорно-двигательного аппарата:
- Остеопороз — уменьшение плотности костной ткани,
- Патология суставов — снижение их подвижности из-за сильной мышечной слабости,
- Сколиоз, кифоз, лордоз — различные формы искривления позвоночника.
Поражение органов пищеварения:
- Запоры — результат гиподинамии,
- Потеря веса из-за разрушения мышц,
- Нарушение процесса жевания и глотания требует питания больного через зонд.
- Поверхностное дыхание,
- Слабый кашлевой рефлекс,
- Частые ОРВИ,
- Недостаток кислорода в крови – головные боли по утрам, пробуждения по ночам, слабость, раздражительность, насыщенные сновидения.
У лиц с миопатией Дюшена развивается кардиомиопатия — слабость миокарда, проявляющаяся повышенной утомляемостью, одышкой, отеками ног, перебоями в работе сердца.
Своевременная диагностика и эффективная терапия могут отсрочить развитие осложнений или совсем предотвратить их появление.
Диагностические мероприятия
Диагностика синдрома Дюшенна не вызывает трудностей у специалистов, поскольку имеет весьма специфические симптомы.
Врачи традиционно начинают с опроса больного или его родителей и сбора анамнестических данных. Особое внимание они уделяют:
- Времени появления первых симптомов,
- Локализации первичной мышечной слабости,
- Общему самочувствию пациента,
- Наличию подобных расстройств у родных и близких.

Во время неврологического обследования выявляется:
- Слабость определенной группы мышц и определяется ее степень,
- Изменение мышечного тонуса,
- Атрофические процессы,
- Гипо- и арефлексия,
- Деформация стопы, груди, позвоночного столба.
Врачи наблюдают за больным ребенком, обращая внимание на то, как он ходит, бегает и встает с пола. Изменение походки — важный диагностический признак миопатии.
После проведения первичных диагностических процедур врачи могут заподозрить наличие у больного патологии и поставить предварительный диагноз. Чтобы его подтвердить или опровергнуть, пациента направляют на лабораторно-инструментальное обследование.
- Анализ крови на гормональный статус.
- Биохимический анализ крови на активность креатинкиназы – фермента, уровень которого у больных детей очень высок. Если КФК в норме, миопатию Дюшена исключают.
- Иммуногистохимическое исследование – микроскопия биоптата мышечной ткани, взятого от больного, с целью определения белка дистрофина. У лиц с миопатией он отсутствует.
- ДНК-тест – генетическое исследование крови больного, позволяющее определить мутантный ген и точно диагностировать патологию.
Дополнительные диагностические методики:
- Электрокардиография — выявление признаков поражения миокарда.
- Электромиография — определение фибрилляции, свидетельствующей о некрозе мышечных волокон. Эта методика оценивает состояние скелетной мускулатуры и подтверждает, что в основе патологии лежит именно поражение мышц, а не нарушение передачи нервных импульсов.
- Дыхательные пробы, рентгенография позвоночника и органов грудной клетки, УЗИ сердца — методы, не оказывающие существенного влияния на процесс постановки диагноза, но позволяющие выявить имеющиеся отклонения в структуре и функционировании органов дыхания и сердца.
Лечебный процесс
Болезнь Дюшенна, как и любая другая наследственная патология, неизлечима. Врачи проводят симптоматическую терапию, устраняющую неприятные проявления, предупреждающую развитие осложнений и продлевающую жизнь больных.

Комплексное поддерживающее лечение болезни:
Профилактика и прогноз
Супругам, в роду которых имелись случаи наследственных заболеваний, перед планированием беременности необходимо посетить врача-генетика. Профилактика патологии также заключается в проведении пренатальной диагностики. Выявив миопатию на ранних сроках, можно прервать беременность.

Миопатия Дюшена — наследственная патология, отличающаяся тяжелым течением и быстрым прогрессированием. Это современная медицинская проблема, характеризующаяся разрушением мышечной ткани и быстрым развитием мышечной слабости. Все без исключения больные погибают в раннем возрасте из-за развития не совместимых с жизнью осложнений. Только адекватная и комплексная терапия, четкое соблюдение рекомендаций врача, тщательный уход и забота родителей могут замедлить ход болезни.
Больные быстро становятся инвалидами и погибают в совсем юные годы. Самое страшное, что детям с миопатией не в силах помочь даже квалифицированные врачи, современные медицинские технологии и терапевтические методики 21 века. Болезнь до сих пор остается неизлечимой, забирая молодые жизни. Современные ученые-медики всего мира трудятся над созданием радикального способа борьбы с миопатией Дюшенна.
Видео: ребенок с синдромом Дюшенна
Читайте также:
