
Миелопатия и арнольда киари
- Описание болезни
- Причины заболевания
- Проявления
- Диагностика
- Лечение
- Видео по теме
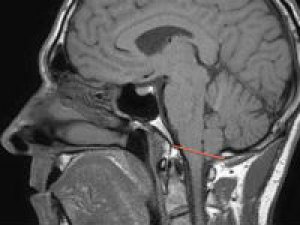
Аномалия Арнольда Киари – нарушение развития, которое заключается в виде несоразмерности размеров черепной ямки и структурных элементов мозга, располагающихся в ней. При этом мозжечковые миндалины спускаются ниже анатомического уровня и могут ущемляться.
Симптомы аномалии Арнольда Киари проявляются в виде частых головокружений, а иногда заканчиваются инсультом мозга. Признаки аномалии могут долго отсутствовать, а затем резко заявить о себе, например, после вирусной инфекции, удара головой или других провоцирующих факторов. Причем случиться это может на любом отрезке жизни.
Описание болезни
Сущность патологии сводится к неправильной локализации продолговатого мозга и мозжечка, в результате чего появляются краниоспинальные синдромы, которые врачи нередко расценивают как атипичный вариант сирингомиелии, рассеянного склероза, спинномозговой опухоли. У большинства больных аномалия развития ромбэнцефалона совмещается с другими нарушениями в спинном мозге – кистами, провоцирующими стремительную деструкцию спинномозговых структур.
Болезнь получила название в честь патологоанатома Арнольда Джулиуса (Германия), который описал аномальное отклонение в конце 18 века и врача из Австрии Ганса Киари, который изучал заболевание в тот же период времени. Распространенность нарушения варьируется в пределах 3–8 случаев на каждые 100000 человек. В основном встречается аномалия Арнольда Киари 1 и 2 степени, а взрослые с 3-м и 4-м типом аномалии живут совсем недолго.
Аномалия Арнольда Киари 1 типа заключается в опускании элементов задней черепной ямки в спинальный канал. Болезнь Киари 2 типа характеризуется изменением местоположения продолговатого мозга и четвертого желудочка, при этом зачастую бывает водянка. Гораздо реже встречается третья степень патологии, которой присущи выраженные смещения всех элементов черепной ямки. Четвертый тип представляет собой дисплазию мозжечка без его сдвига вниз.
Причины заболевания
По данным ряда авторов, болезнь Киари представляет собой недоразвитие мозжечка, сочетающееся с различными отклонениями в отделах мозга. Аномалия Арнольда Киари 1 степени – наиболее распространенная форма. Это нарушение представляет собой одностороннее или двухстороннее опускание миндалин мозжечка в спинальный канал. Это может произойти вследствие перемещения продолговатого мозга вниз, часто патология сопровождается различными нарушениями краниовертебральной границы.
Клинические проявления могут возникнуть только на 3–4 десятке жизни. При этом следует отметить, что бессимптомное течение эктопии миндалин мозжечка в лечении не нуждается и часто проявляется случайно на МРТ. На сегодняшний день этиология болезни, так же как и патогенез, изучены плохо. Определенная роль отводится генетическому фактору.
Выделяют три звена в механизме развития:
- генетически обусловленная врожденная остеоневропатия;
- травматизация ската во время родов;
- высокое давление ликвора на стенки спинномозгового канала.
Проявления
По частоте возникновения выделяют следующие симптомы:
- головные боли – у трети пациентов;
- боль в конечностях – 11%;
- слабость в руках и ногах (в одной или двух конечностях) – больше половины пациентов;
- чувство онемения в конечности – половина больных;
- снижение или утрата температурной и болевой восприимчивости – 40%;
- шаткость походки – 40%;
- непроизвольные колебания глаз – треть больных;
- двоение в глазах – 13%;
- нарушения глотания – 8%;
- рвота – у 5%;
- нарушения произношения – 4%;
- головокружения, глухота, онемение в лицевой области – у 3% больных;
- синкопальные (обморочные) состояния – 2%.
Болезнь Киари второй степени (диагностируется у детей) сочетает в себе дислокацию мозжечка, ствола и четвертого желудочка. Неотъемлемый признак – наличие менингомиелоцеле в области поясницы (грыжа спинального канала с выпячиванием вещества спинного мозга). Неврологическая симптоматика развивается на фоне аномального строения затылочной кости и шейного отдела позвоночного столба. Во всех случаях присутствует гидроцефалия, часто – сужение водопровода мозга. Неврологические признаки появляются с самого рождения.
Операция при менингомиелоцеле проводится в первые дни после рождения. Последующее хирургическое расширение задней черепной ямки позволяет добиться хороших результатов. Многие пациенты нуждаются в шунтировании, особенно при стенозе Сильвиевого водопровода. При аномалии третьей степени черепно-мозговая грыжа внизу затылка или в верхней шейной области сочетается с нарушениями развития мозгового ствола, краниального основания и верхних позвонков шеи. Образование захватывает мозжечок и в 50% случаев – затылочную долю.
Эта патология встречается очень редко, имеет неблагоприятный прогноз и резко сокращает продолжительность жизни даже после операции. Сколько именно человек будет жить после своевременного вмешательства, точно сказать нельзя, но, вероятнее всего, что недолго, так эта патология считается несовместимой с жизнью. Четвертая степень заболевания представляет собой обособленную гипоплазию мозжечка и на сегодняшний день не относится к симптомокомплексам Арнольда-Киари.
Клинические проявления при первом типе прогрессируют медленно, в течение нескольких лет и сопровождаются включением в процесс верхнего шейного спинномозгового отдела и дистального отдела продолговатого мозга с нарушением работы мозжечка и каудальной группы черепных нервов. Таким образом, у лиц с аномалией Арнольда-Киари выделяют три неврологических синдрома:
- Бульбарный синдром сопровождается дисфункцией тройничного, лицевого, преддверно-улиткового, подъязычного и вагусного нервов. При этом наблюдаются нарушения глотания и речи, бьющий вниз спонтанный нистагм, головокружения, расстройства дыхания, парез мягкого неба с одной стороны, охриплость голоса, атаксии, дискоординация движений, неполный паралич нижних конечностей.
- Сирингомиелитический синдром проявляется атрофией мышц языка, нарушением глотания, отсутствием чувствительности в лицевой области, хриплостью голоса, нистагмом, слабостью в руках и ногах, спастическим повышением мышечного тонуса и т. д.
- Пирамидный синдром характеризуется незначительным спастическим парезом всех конечностей с гипотонусом рук и ног. Сухожильные рефлексы на конечностях повышаются, брюшные рефлексы не вызываются или снижаются.
Боли в области затылка и шеи могут усиливаться при покашливании, чихании. В руках снижается температурная и болевая чувствительность, а также мышечная сила. Часто возникают обмороки, головокружения, у больных ухудшается зрение. При запущенной форме появляются апноэ (кратковременная остановка дыхания), быстрые неконтролируемые движения глаз, ухудшение глоточного рефлекса.
Интересный клинический признак у таких людей – провоцирование симптомов (синкопе, парестезии, боли и др.) натуживанием, смехом, кашлем, пробой Вальсальвы (усиленный выдох при закрытом носе и рте). При нарастании очаговых симптомов (стволовых, мозжечковых, спинномозговых) и гидроцефалии встает вопрос о хирургическом расширении задней черепной ямки (субокципитальной декомпрессии).
Диагностика
Диагноз аномалии первого типа не сопровождается повреждением спинного мозга и ставится в основном у взрослых посредством КТ и МРТ. По данным патологоанатомического вскрытия, у детей с грыжей спинномозгового канала болезнь Киари второго типа выявляют в большинстве случаев (96–100%). С помощью УЗИ можно определить нарушения циркуляции ликвора. В норме цереброспинальная жидкость легко циркулирует в подпаутинном пространстве.
Боковой рентген и МР картина черепа отображает расширение канала позвоночного столба на уровне С1 и С2. На ангиографии сонных артерий наблюдается огибание миндалины мозжечковой артерией. На рентгене отмечаются такие сопутствующие изменения краниовертебральной области, как недоразвитие атланта, зубовидного отростка эпистрофея, укорачивание атлантозатылочной дистанции.
При сирингомиелии на боковом снимке рентгена наблюдается недоразвитие задней дуги атланта, недоразвитие второго шейного позвонка, деформация большого затылочного отверстия, гипоплазия боковых частей атланта, расширение позвоночного канала на уровне С1-С2. Дополнительно следует провести МРТ и инвазивное рентгенологическое исследование.
Манифестация симптомов болезни у взрослых и лиц пожилого возраста часто становится поводом для выявления опухолей задней черепной ямки или краниоспинальной области. В некоторых случаях правильно поставить диагноз помогают имеющиеся у пациентов внешние проявления: низкая линия оволосения, укороченная шея и др., а также наличие на рентгене, КТ и МРТ краниоспинальных признаков костных изменений.
Для уточнения диагноза используют различные плоскости сканирования, благодаря чему можно обнаружить несколько информативных в отношении болезни симптомов у плода. Получить изображение во время беременности достаточно легко. Ввиду этого УЗИ остается одним из основных вариантов сканирования для исключения патологии у плода во втором и третьем триместрах.
Лечение
При бессимптомном течении показано постоянное наблюдение с регулярным ультразвуковым и рентгенографическим исследованием. Если единственный признак аномалии – незначительные боли, пациенту назначают консервативное лечение. Оно включает разнообразные варианты с использованием нестероидных противовоспалительных средств и миорелаксантов. К наиболее распространенным НПВС относятся Ибупрофен и Диклофенак.
Нельзя самостоятельно назначать себе обезболивающие препараты, так как они имеют ряд противопоказаний (например, язвенная болезнь). При наличии какого-либо противопоказания врач подберет альтернативный вариант лечения. Время от времени назначают дегидратационную терапию. Если в течение двух-трех месяцев эффекта от такого лечения нет, проводят операцию (расширение затылочного отверстия, удаление дужки позвонка и т. д.). В этом случае требуется строго индивидуальный подход, позволяющий избежать как ненужного вмешательства, так и проволочки с операцией.
У некоторых пациентов хирургическая ревизия является способом постановки конечного диагноза. Цель вмешательства – ликвидация сдавливания нервных структур и нормализация ликвородинамики. Такое лечение приводит к существенному улучшению у двух-трех пациентов. Расширение черепной ямки способствует исчезновению головных болей, восстановлению осязаемости и подвижности.
Благоприятный прогностический признак – расположение мозжечка выше С1 позвонка и наличие только мозжечковой симптоматики. В течение трех лет после вмешательства могут возникать рецидивы. Таким пациентам по решению медико-социальной комиссии присваивается инвалидность.
Общая информация
Синдром Арнольда-Киари представляет собой набор признаков и симптомов, вызванных редкой мальформацией (отклонение от нормального развития, аномалия) задней черепной ямки; у пострадавших эта структура развита слабо, поэтому мозжечок выходит (выступает) из своего естественного участка через затылочное отверстие, расположенное у основания черепа.
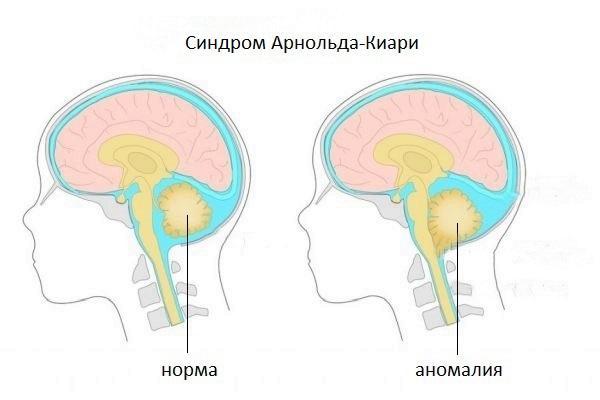
Есть четыре различных типа синдрома Арнольда-Киари; особенность, отличающая один тип от другого, является степень выпячивания, следовательно, доля вовлеченного материала мозжечка. Тип I является наименее тяжелым (иногда остается бессимптомным на протяжении всей жизни), тогда как IV тип наиболее тяжелый; однако уже со второго типа качество жизни больного ставится под угрозу.
Симптомы, характеризующие аномалии Арнольда-Киари многочисленны и варьируются от головных болей до слабости мышц и проч.
На сегодняшний день не существует лекарств, позволяющих устранить порок развития мозжечка, однако существуют способы лечения, позволяющие частично смягчить симптомы.
Что такое синдром Арнольда-Киари?
Синдром Арнольда-Киари, или мальформация Арнольда-Киари — структурное изменение мозжечка, характеризующееся его смещением вниз, именно в направлении позвоночного канала и затылочного отверстия, базальные части полушария мозжечка.
Простыми словами, это грыжа мозжечка, при которой часть мозжечка выступает из затылочного отверстия, проникая в позвоночный канал.
Синдром Арнольда-Киари получил свое название от двух врачей, которые впервые описали его, Арнольда Джулиуса и Ганса Киари.
Причины и факторы риска
Исследователи полагают, что синдром Арнольда-Киари может иметь наследственное происхождение, так как обнаруживалась среди членов одной семьи. Тем не менее, генетические условия, вызывающие заболевание (т.е., какие и сколько генов участвуют) и тип передачи еще предстоит выяснить.
Исходя из серьезности выпячивания и момента жизни, в котором он возникает, заболевание можно разделить на 4 различных типа, идентифицированных первыми четырьмя римскими числами (I, II, III и IV).
Первые два типа по сравнению со вторыми более распространены и менее серьезны; Тип III и тип IV, на самом деле, очень редки и несовместимы с жизнью.
— Мальформация I типа.
Первая степень синдрома протекает бессимптомно (т.е. без явных симптомов), по крайней мере, до конца детства или юности.
Причина его возникновения кроется в уменьшенном черепном пространстве: в таких условиях часть мозжечка (именно миндалина(и), расположенная(ые) с нижней стороны), из-за недостатка места, вынуждена проникать в затылочное отверстие и входить в позвоночный канал.
Примечание: у некоторых взрослых людей с синдром Арнольда-Киари 1 типа все в порядке и они ведут совершенно нормальную жизнь. Это связано с тем, что аномалия мозжечка не настолько серьезна, чтобы вызывать симптомы или нарушения. Поэтому очень часто эти субъекты игнорируют свое состояние или узнают о нем по чистой случайности.
— Мальформация II типа.
2 тип мальформации Арнольд-Киари является врожденным заболеванием, которое присутствует с рождения ребенка, и всегда протекает симптоматически.
По сравнению с 1 степенью он характеризуется большим выпячиванием черепной ямки, при котором помимо миндалин мозжечка также выпячивает часть мозжечка (называемая червь мозжечка) и венозный сосуд.
Почти всегда мальформация Арнольд-Киари II типа ассоциируется с особой формой расщелины позвоночника, называемой миеломенингоцеле.
Среди различных последствий этой аномалии выделают: блокирование потока ликвора (спинномозговой жидкости) через затылочное отверстие (что приводит к состоянию, называемому гидроцефалией) и прерывание нервных сигналов.
Первоначально термин Арнольд-Киари относился только ко 2 типу заболевания. Теперь, её обычно используют для всех форм болезни.
— Мальформация III типа.
Присутствующий с рождения, III тип порока вызывает серьезные неврологические проблемы, настолько, что часто несовместимы с жизнью. В этих случаях на самом деле наблюдается выпячивание мозжечка, и по этой причине говорится о затылочном энцефалоцеле.
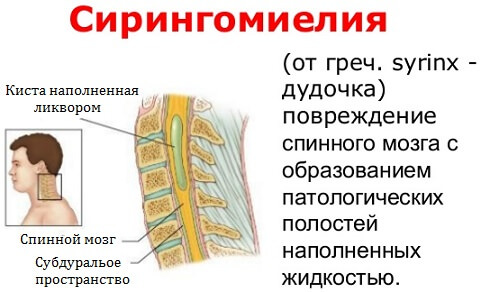
Обычно III тип состояния характеризуется гидроцефалией и сирингомиелией; последний представляет собой особое состояние, характеризующееся наличием одной или нескольких кист в позвоночном канале.
— Мальформация IV типа.
Мальформация Арнольда-Киари IV типа характеризуется отсутствием развития части мозжечка (недоразвитие мозжечка).
Аномалия врожденная и абсолютно несовместима с жизнью.
Врачи и ученые отметили, что следующие заболевания являются частыми среди людей с мальформацией Киари:
Точная частота пороков развития неизвестна; это связано с тем, что у некоторых даже взрослых людей с I типом мальформации Арнольда-Киари никаких симптомов нет, и они кажутся совершенно нормальными (поэтому болезнь недиагностируется).
Несколько достоверных эпидемиологических исследований сообщают, что:
- I тип симптоматичен у 1 из 100 детей;
- II тип особенно широко распространен в популяциях кельтского происхождения;
- женщины страдают в 3 раза чаще, чем мужчины.
Симптомы и осложнения

4 типа заболевания имеют разные симптомы и признаки.
Ниже приводится таблица с точным описанием симптомов, которые характеризуют I, II и III типы синдрома.
Для IV типа невозможно проследить симптоматику, так как это состояние неизбежно и внезапно приводит к гибели плода.
Расщелина позвоночника (миеломенингоцеле).
Расщелина позвоночника — врожденный порок развития позвоночника, из-за которого менинги, а иногда и спинной мозг выходят из своего места (обычно они ограничены позвонками). Миеломенингоцеле является наиболее тяжелой формой расщелины позвоночника: у пораженных выпячивают менинги и спинной мозг из позвоночной камеры и образуют мешок на уровне спины. Эта сумка, хотя и защищена слоем кожи, подвержена внешним воздействиям и постоянно подвергается риску серьезных, а в некоторых случаях даже смертельных инфекций.
Синдром Арнольда-Киари типа II, III и IV видны уже в пренатальном возрасте (т.е., когда пораженный ребенок все еще находится в материнской утробе) при ультразвуковом исследовании.
Что касается I типа, желательно обратиться к врачу, как только появятся типичные симптомы, о которых говорилось выше. Важно также проходить своевременные обследования, так как в результате последних могут возникнуть другие сопутствующие нарушения.
Осложнения
Осложнения синдром Арнольда-Киари связаны с ухудшением выпячивания мозжечка или патологическими состояниями, связанными, следовательно, с гидроцефалией, миеломенингоцеле, сирингомиелией и проч.
Ухудшение выпячивания (протрузии), обусловленное повышенным давлением черепа на мозжечок, очевидно, предполагает обострение симптомов.
Диагностика
Диагностические тесты, которые позволяют установить степень протрузии мозжечка через затылочное отверстие (таким образом, устанавливая тип мальформации Арнольда-Киари):
- Магнитно-резонансная томография (МРТ). Благодаря формированию магнитных полей, он позволяет получить детальное изображение мозжечка и позвоночного канала, не подвергая пациента вредному ионизирующему излучению.
- Компьютерная томография (КТ) дает четкие изображения внутренних органов, в том числе мозжечка и спинного мозга. Во время его выполнения субъект подвергается минимальному воздействию вредного ионизирующего излучения.
КТ и МРТ, которым предшествует точное физическое обследование, имеют основополагающее значение для выявления любых патологий, связанных с синдромом Арнольда-Киари.
Таблица. Как и когда диагностируется аномалия Арнольда-Киари развития заболевания.
| Тип порока развития | Когда и как это можно диагностировать? |
| I | В позднем детстве или позднем подростковом возрасте посредством объективных обследований с последующим КТ и/МРТ. |
| II | В дородовом возрасте при УЗИ. При рождении и в раннем детстве, посредством объективных обследований, компьютерной томографией и/или МРТ. |
| III | В дородовом возрасте при УЗИ. После рождения и в раннем детстве, посредством объективных обследований, компьютерной томографией и/или МРТ. |
| IV | В дородовом возрасте при УЗИ. |
Лечение
Синдром Арнольда-Киари неизлечим. Однако существуют как фармакологические, так и хирургические методы терапии, позволяющие частично смягчить признаки заболевания.
— Медикаментозная терапия.
Пациенты с мальформацией Арнольда-Киари I типа, страдающий от головной боли и боли в шее и/или лице, могут принимать обезболивающие препараты.
Выбор наиболее подходящих лекарств для конкретного случая остается за лечащим врачом.
— Хирургическое лечение.
Цель хирургического лечения состоит в том, чтобы уменьшить давление, оказываемое черепом, чтобы предотвратить повреждение мозжечка и спинного мозга.
Для достижения этой цели есть несколько процедур, таких как:
- Декомпрессия задней черепной ямки, во время которой хирург удаляет часть задней части затылочной кости.
- Декомпрессия спинного мозга с помощью ламинэктомии (декомпрессионная ламинэктомия). Во время его выполнения хирург удаляет пластинку второго и третьего шейного позвонка. Пластинка — это позвоночная часть, отделяющая отверстие, через которое проходит спинной мозг..
Примечание: иногда декомпрессия задней ямки и декомпрессивная ламинэктомия выполняются одновременно. - Декомпрессионный разрез твердой мозговой оболочки. При разрезе твердой мозговой оболочки или наружного менинга пространство, доступное мозжечку, увеличивается, а давление на его повреждение уменьшается. Чтобы покрыть и защитить трещину, созданную разрезом, хирург пришивает на нее кусок искусственной ткани (или взятый из другой части тела).
- Хирургическое шунтирование (создание дополнительного пути в обход пораженного участка). Это, по сути, дренажная система, состоящая из гибкой трубки, позволяющей удалять спинномозговую жидкость, в случае гидроцефалии, или опорожнять цисту(ы), в случае сирингомиелии. Не исключено, что пациенты с гидроцефалией будут вынуждены проходить хирургическим шунтом всю жизнь.
— Осложнения хирургического вмешательства.
Риски, связанные с хирургией, различны. Фактически возможно появление:
- кровотечений;
- повреждение структур головного мозга и/или спинного мозга;
- инфекционный менингит;
- проблемы с заживлением ран;
- необычные скопления жидкости вокруг мозжечка.
Помните, что любое повреждение головного или спинного мозга, произошедшее во время операции, непоправимо. Поэтому, прежде чем подвергнуться больного вмешательству любого типа, лечащий врач выявит любые риски и осложнения необходимой процедуры.
Прогноз
Синдром Арнольда-Киари типа II, III и IV никогда не имеют положительного прогноза, поскольку, помимо того, что неизлечимы, могут вызывать серьезные неврологические нарушения или даже быть несовместимыми с жизнью.
Прогноз для пациентов с I типом часто неизвестен. Многие люди с этим заболеванием не имеют никаких симптомов, и невозможно предсказать, будут ли симптомы развиваться в будущем. Другие люди с мальформацией Арнольда-Киари могут испытывать головокружение, мышечную слабость, онемение, проблемы со зрением, головную боль или проблемы с равновесием и координацией. У этих людей не всегда возможно предсказать, будут ли симптомы ухудшаться с течением времени.
Людям с пороком 1 типа важно регулярно проходить медицинские обследования, чтобы быть под наблюдением врача при появлении любых новых симптомов.

Аномалия Арнольда-Киари — это отклонение в развитии. И конкретно отклонение касается связки головного и спинного мозга. Головной мозг переходит в спинной на уровне большого затылочного отверстия. Четкой границы перехода нет.
Однако расположение ствола мозга относительно костей черепа и шейного отдела позвоночника по разным причинам может меняться. Это несовпадение приводит к сжатию спинного мозга в районе шейного отдела. Нормальная циркуляция спинномозговой жидкости нарушается.
Эта ситуация еще называется мальформация. Название происходит от латинских malus (что означает плохой) и formation (образование). Проще говоря возникает аномалия развития, за которой стоят изменение строения и функций.
В данном случае имеет место мальформация Арнольда-Киари в месте перехода черепа в позвоночник, то есть мальформация головного мозга.
Описали эту патологию австрийский патологоанатом Ханс Киари (в 1891 году) и немецкий патологоанатом Юлиус Арнольд (в 1894 году). Отсюда и сложное название.
Статистика указывает на то, что частота заболевания не такая уж и редкая — до 8.4 заболевших на 100 тысяч населения. Дополнительно к аномалии (до 80% случаев) выявляется сирингомиелия (образование кист в ткани спинного мозга).
Аномалия Арнольда-Киари — что это
Справочно. Аномалия Арнольда-Киари — это нарушение развития, которое приводит к опущению мозжечковых миндалин ниже анатомического уровня и к ущемлению продолговатого мозга.
Нормой считается расположение миндалин мозжечка выше большого затылочного отверстия. В процессе мальформации они могут сместиться даже до уровня второго шейного позвонка. При таком смещении усиливается блокировка тока спинномозговой жидкости.
Не всегда удается распознать это заболевание сразу, а затем происходит его резкая манифестация. Проявление патологии происходит к 25 — 40 годам.
При обнаружении характерных для аномалии симптомов необходимо обратиться к врачу, в противном случае риск развития инфаркта спинного мозга значительно возрастает.
Поскольку речь идет об отклонении от нормального развития организма, заболевание часто называют мальформация Арнольда-Киари.
Особенности мальформации
Справочно. Задний отдел мозга перемещается к большому затылочному отверстию (БЗО), когда у черепной ямки оказываются слишком малые размеры. В итоге формируется порок, возникают нарушения циркуляции ликвора.
Кости черепа не позволяют БЗО менять свой диаметр, поэтому при любых смещениях мозговых структур ущемляются близлежащие ткани. Последствия такого явления могут носить для пациента фатальный характер.
Продолговатый мозг отвечает за работу сердечно-сосудистой и дыхательной системы организма. Его сдавливание приводит не только к неврологическому дефициту, но может давать более опасные последствия, вплоть до смерти больного.
Смещение правого и левого полушарий мозжечка останавливает движение ликвора, что провоцирует гидроцефалию. Водянка повышает риск осложнений тех расстройств, которые уже имеются у пациента.
Процент пациентов с врожденной формой аномалии Киари невелик.
Справочно. Последние данные говорят о приобретенном характере. Дистопия миндалин мозжечка обусловлена быстрым ростом тканей при медленно протекающих изменениях в строении черепа. В медицине также известна под названием эктопия миндалин мозжечка.
Синдром Арнольда-Киари без проявления симптоматики может случайно обнаружиться во время проведения МРТ.
Причины развития аномалии
Мнения медиков о причинах появления аномалии расходятся. Есть несколько теорий, объясняющих то, каким образом развивается порок.
Неврологи выделяют две патологии, приводящие к формированию мальформации Киари (Chiari malformation):
- Развитие плода в утробе матери может пойти с нарушением — черепная ямка окажется меньше анатомической нормы, отделы мозга приобретут обычные параметры.
- Размеры отделов увеличены, при этом параметры задней черепной ямки и БЗО отвечают нормам. Увеличивающийся мозг устремляется к отверстию.
Врожденная аномалия у ребенка развивается, если беременная женщина не контролирует прием лекарств, употребляет алкоголь, курит на ранних сроках беременности. 
Кроме того, вирусные инфекции у будущей матери (краснуха, цитомегаловирус) могут пагубно сказаться на развитии плода.
Справочно. Возникновению заболевания могут предшествовать различные родовые травмы, гидроцефалия, сильные повреждения головы у взрослого человека.
Типы аномалии
Рассматриваются четыре типа. Классификация производится с ориентацией на определенные основания.
Существенными признаками оказываются следующие изменения: те, что произошли в головном мозге на структурном уровне, те, что говорят о недоразвитости черепной коробки.
Аномалия Арнольда-Киари 1 типа отличается сдвигом мозжечковых миндалин, сопровождается нарушением циркуляции ликвора.
Последний заполняет узкий канал спинного мозга, вызывая гидромиелию. Указанный тип аномалии носит благоприятный прогноз. Он часто диагностируется у подростковой и взрослой групп населения.
Аномалия Арнольда-Киари 2 типа проявляет себя у новорожденных детей. Здесь наблюдается еще большее смещение отделов. Помимо мальформации, у грудничков диагностируется спинномозговая грыжа, обнаруживается аномальное развитие позвоночного столба.
В области затылка происходит выпячивание мозгового вещества через мягкую оболочку, мозжечок оказывается там же. Такова картина аномалии Арнольда-Киари 3 типа.
Внимание. Аномалия Арнольда-Киари 4 типа дает о себе знать тем, что мозжечок новорожденного оказывается недоразвитым, не занимает должного анатомического положения. Такая патология делает младенца неприспособленным к жизни, летальный исход неизбежен.
Степени тяжести
Сколько живут с аномалией Арнольда-Киари 1-й степени? Такой вопрос часто задают люди, услышавшие свой диагноз. Такая степень тяжести является самой невысокой, клинические проявления могут не отмечаться.
Спровоцировать возникновение симптомов могут черепно-мозговые травмы и повреждения позвоночника в верхней его части. Также запустить процесс может развившаяся нейроинфекция.
Аномалии 2 и 3-й степени уже сопровождаются патологическими изменениями в нервной ткани. У больного часто обнаруживают:
- смещение мозгового вещества;
- кисты проводящих ликвор путей;
- недоразвитость некоторых извилин мозга;
- гипоплазию подкорковых узлов.
Аномалия Арнольда-Киари — симптомы
Говоря о симптомах аномалии, надо, прежде всего, различать вариационные ее типы. Первый тип мальформации сопровождается несколькими синдромами, среди которых: гипертензионный, церебеллярный, сирингомиелический, бульбарный и т.д.
Гипертензионный синдром представляет собой повышение давления внутри черепа (ВЧД). Характерными симптомами будут интенсивные затылочные боли, тошнота, рвота, ригидность шейных мышц.
Церебеллярный синдром характеризуется речевыми расстройствами, нарушениями двигательной функции. При этом отсутствует четкость движений, затруднена мелкая моторика.
Сирингомиелический синдром проявляется потерей чувствительности в конечностях. Больной может получить случайный ожог, не заметив этого сразу. При обследовании обнаруживаются кисты спинного мозга.
Другие типы аномалии сопровождаются более тяжелой симптоматикой.
Справочно. У новорожденных страдает дыхание, возможна его остановка, отмечаются нарушения в глотании. Ребенок не может полноценно питаться. Посинение кожных покровов, гипертонус мышц, нистагм — вот основные проявления аномалии этих типов.
Возможные осложнения
Мальформация в некоторых случаях провоцирует достаточно опасные осложнения и может привести многих пациентов к инвалидности. Часто отмечаются увеличение ВЧД, дыхательные расстройства, апноэ, на фоне мальформации развиваются инфекционные заболевания легких и мочеполовой системы.
Справочно. Тяжело протекающая патология может стать причиной наступления комы, остановки в работе сердца, в итоге, быстрой смерти.
В запущенных случаях реанимация позволяет лишь поддерживать жизненно важные функции, сдавленный мозг восстановить практически невозможно.
Постановка диагноза
Осмотр невролога, сбор анамнеза представляют собой часть диагностики — они необходимы, но недостаточны.
Энцефалограмма, диагностика нарушения кровообращения в головном мозге и шейном отделе позвоночника могут косвенно показать наличие ВЧД. 
С помощью рентгенографии, компьютерной томографии можно зафиксировать дефекты формирования черепной коробки. Но для определения состояния нервной ткани такие способы окажутся малоинформативными.
Справочно. МРТ сегодня считается единственно достоверным методом, позволяющим провести точную диагностику и своевременно распознать синдром Арнольда-Киари.
Процедура предполагает полное обездвиживание пациента, чего легко добиться от взрослого человека. Трудности возникают, когда пациентами являются маленькие дети. В этом случае необходимо применение общего наркоза.
Варианты лечения
После постановки диагноза лечение больного осуществляет нейрохирург или невролог. В исключительных случаях устранение аномалии возможно лишь путем проведения операции.
Если единственный симптом болезни — головная боль, то врачи ограничиваются медикаментозной терапией. Специалисты подбирают препараты:
- устраняющие воспаление (Найз, Ибупрофен, Диклофенак);
- анальгетики (Кеторол);
- спазмолитики (Мидокалм).
Справочно. Показанием к операции является сильное сдавливание отделов мозга, явно выраженные неврологические расстройства, если положительный эффект от приема лекарственных препаратов не наблюдается.
Благодаря хирургическому вмешательству можно устранить чрезмерное давление на нервную ткань и привести в норму движение ликвора. Одной из проводимых операций является краниовертебральная декомпрессия, направленная на увеличение размеров задней черепной ямки.
Внимание. Декомпрессия относится к классу травматичных и рискованных операций. Согласно статистике, она приводит к осложнениям у каждого десятого пациента.
Риск летального исхода у прооперированных больных выше, чем у тех, кто операцию не переносил. Нейрохирурги стараются проводить такой тип вмешательства только в самых крайних случаях, когда налицо явные признаки сдавливания мозга. 
Иной вариант устранения последствий мальформации предполагает шунтирование, которое способно обеспечить отвод ликвора из черепной коробки. Благодаря имплантации специальных трубок жидкость перетекает в грудную и брюшную полости, ВЧД снижается.
В наиболее острых случаях требуется немедленная госпитализация больного и проведение всего спектра терапевтических, профилактических и коррекционных процедур.
Прогноз выживаемости
Продолжительность жизни зависит от типа аномалии и степени ее тяжести. Первый тип позволяет сделать благоприятный прогноз, поскольку симптоматика либо вовсе отсутствует, либо возникает после получения травм головы.
Если никаких проявлений болезни нет, то продолжительность жизни больных такая же, как и у здоровых людей.
Для пациентов со вторым типом аномалии прогноз хуже, она переносится тяжелее.
Иногда борьба с очаговой неврологической симптоматикой не приносит плодов даже при активном лечении медикаментами. В таком случае требуется хирургическое вмешательство, чтобы впоследствии изменения по неврологической части были менее выраженными.
Справочно. Третий и четвертый типы мальформации являются самыми тяжелыми для пациентов, прогноз зачастую неблагоприятен.
Заболевание затрагивает важные структуры мозга, у больного отмечаются пороки внутренних органов. Часто функции ствола мозга страдают настолько, что нарушения оказываются несовместимыми с жизнью.
Читайте также:
