
Что за заболевание врожденная мышечная дистрофия
Мышечные дистрофии представляют собой группу наследственных первичных заболеваний мышц, характеризующихся дегенерацией мышечных волокон и мышечной слабостью.
Мышечная слабость при мышечной дистрофии
Классификация этих состояний традиционно основывалась на:
- патологических,
- клинических,
- наследственных паттернах.
Даже при том, что есть некоторые общие признаки, относящиеся к мышечной слабости, определенные синдромы показывают некоторые характерные клинические проявления.
Мышечная слабость, возникающая при всех различных типах мышечной дистрофии, имеет определенную характеристику. Прежде всего, пациент ощущает слабость с обеих сторон тела. Как правило, при мышечной слабости отсутствуют болевые ощущения, но мышцы чувствительны к прикосновениям. Известно, что при некоторых типах дистрофий, затрагивающих основные мышцы конечностей, возникают скованность и судороги (хотя тяжелые формы судорог довольно необычны).
Течение и степень тяжести заболевания, а также его прогноз зависят от конкретного типа дистрофии, от которой страдает человек, следовательно, необходим точный диагноз. Тщательное изучение истории болезни и соответствующие лабораторные анализы имеют решающее значение для установления диагноза и планирования терапевтического процесса.
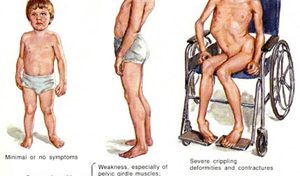
Симптомы мышечной дистрофии Дюшенна обычно появляются в возрасте до 6 лет. Слабость начинается в области верхних конечностей и таза. Дети, страдающие данным заболеванием, часто падают, имеют проблемы с двигательными навыками (бег, прыжки) и проявляют псевдогипертрофию. Симптомы могут также включать усталость, трудности в обучении (IQ может быть ниже 75) и возможную умственную отсталость.

Мышечная дистрофия Беккера менее серьезна, чем мышечная дистрофия Дюшенна, поэтому основное различие заключается в том, что люди с этим заболеванием могут ходить в 16 лет (а некоторые продолжают делать это и в пожилом возрасте).
У них могут быть:
- судороги в мышцах,
- определенные трудности с координацией.
Наиболее выраженным симптомом миотонической мышечной дистрофии является неспособность расслабить мышцы после внезапного сокращения. Это состояние поражает как женщин, так и мужчин в возрасте от 20 до 30 лет.
Другие клинические проявления включают:
- проблемы со зрением,
- трудности с глотанием,
- сильное снижение веса,
- облысение,
- сердечные заболевания,
- атрофию яичек.
Термин "врожденная мышечная дистрофия" используется для группы мышечных дистрофий, объединенных тем фактом, что мышечная слабость начинается в младенчестве или в раннем детстве (обычно в возрасте до двух лет). Из-за этих состояний ребенок может казаться "дискетным", и позже дети медленно достигают таких основных двигательных показателей, как сидение, переворачивание или ходьба. Некоторые из более редких форм врожденной мышечной дистрофии также сопровождаются умственной отсталостью или неспособностью к обучению.
Ранние признаки мышечной дистрофии Эмери-Дрейфуса включают ходьбу на пальцах ног из-за жесткости ахиллова сухожилия в пятках, трудности сгибания в локтях и возможность обморока из-за аномалий сердца.
Мышечная дистрофия конечностей, как и другие мышечные дистрофии, в первую очередь представляет собой расстройство произвольных мышц. Люди с этим заболеванием сталкиваются со слабостью мышц бедер и ног.
Встройте "Правду.Ру" в свой информационный поток, если хотите получать оперативные комментарии и новости:
Подпишитесь на наш канал в Яндекс.Дзен или в Яндекс.Чат
Добавьте "Правду.Ру" в свои источники в Яндекс.Новости или News.Google
Также будем рады вам в наших сообществах во ВКонтакте, Фейсбуке, Твиттере, Одноклассниках.

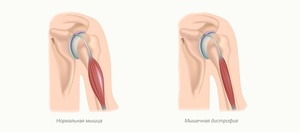
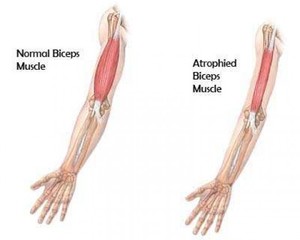
Мышечная дистрофия (МД) — это группа заболеваний, характеризующихся прогрессирующей слабостью и мышечной дегенерацией. Мышцы постепенно атрофируются — теряют свой объем и, следовательно, силу.
Это болезни генетического происхождения, которые могут возникать в любом возрасте: с самого рождения, в детстве или во взрослой жизни. Существует более 30 форм заболеваний, которые различаются по возрасту появления симптомов, характеру пораженных мышц и степени тяжести. Большинство типов дистрофий постепенно осложняются и имеют необратимые последствия. В настоящее время лечения МД все еще не существует. Наиболее известным и распространенным типом заболеваний является миопатия Дюшенна.
В ходе развития МД страдают в первую очередь основные мышцы, которые способствуют произвольному движению, включая мышцы, бедра, ног, рук и предплечья. В некоторых случаях могут быть затронуты респираторные мышцы и сердце. Люди с мускульной дистрофией постепенно теряют свою мобильность при ходьбе. Другие симптомы могут быть связаны с мышечной слабостью, включая сердце, желудочно-кишечные, глазные проблемы.
Распространенность заболевания
Миопатия относится к редким и неизлечимым заболеваниям. Трудно вывести точную статистику, поскольку она объединяет различные болезни. Согласно некоторым исследованиям, около 1 из 3 500 человек страдают от этого заболевания.
Например:
![]()
Миопатия Дюшенна затрагивает приблизительно одного ребенка (мальчика) из 3500.- Миопатия Беккера касается 1 мальчика из 18 000.
- Фазио-скапулогумаральная дистрофия поражает около 1 из 20 000 взрослых людей.
- Болезнь Эмери-Дрейфус затрагивает 1 из 300 000 человек, вызывает ретракцию сухожилия и нарушение сердечной мышцы
Частота и тип заболеваний зависит от конкретной страны:
Причины заболевания и лечение
Причиной данной патологии являются генетические заболевания, то есть дефект (или мутация) гена, необходимого для нормального развития мышц. Когда этот ген мутирует, мышцы больше не в состоянии нормально функционировать — они теряют свой силовой потенциал и в результате атрофируются.
Например:
- Миопатия Дюшенна связана с дефицитом дистрофина — белка, расположенного под мембраной мышечных клеток, который играет роль в сокращении мышц.
- Почти у половины врожденных МД причиной является дефицит мерозина — белка, составляющего мембрану клеток мышцы.
Как правило, МД передается рецессивно. Другими словами, для того чтобы болезнь выражалась, оба родителя должны быть носителями и передавать ребенку ненормальный ген. Болезнь не проявляется у родителей по той причине, что у каждого из них есть только один аномальный ген, а не два. Для нормального функционирования мышц достаточно одного нормального гена.
Кроме того, некоторые формы миопатии затрагивают только мальчиков: это миопатия Дюшенна и Беккера. В обоих случаях ген, участвующий в этих двух заболеваниях, расположен в Х-хромосоме, которая существует в единственной копии у мужского пола.
Симптомы заболевания
МД проявляются мышечной слабостью, которая имеет тенденцию к постепенному ухудшению, симптомы варьируются в зависимости от типа патологии. В зависимости от случая могут присутствовать и другие симптомы, такие как сердечные и респираторные расстройства, аномалии глаз (пороки развития, катаракта), интеллектуальный дефицит, гормональные нарушения и т. д.
Дети часто жалуются на судороги и мышечные боли. Болезнь развивается довольно быстро, как только появляются первые симптомы. Обычно использование инвалидной коляски требуется примерно в возрасте 12 лет. Такого рода нарушения приводят к сколиозу и деформациям суставов. Кроме того, у некоторых детей наблюдается умственная отсталость. К концу подросткового возраста часто возникают сердечные осложнения (сердечная недостаточность), а также респираторные проблемы, требующие искусственной подачи воздуха. Средняя продолжительность жизни (от 20 до 30 лет в среднем).
Миопатия Беккера. Симптомы сравнимы с симптомами М. Д. Дюшенна , однако они менее выражены, а развитие заболевания происходит медленнее. Симптомы начинаются в 5−15 лет, иногда позже, характеризуются прогрессирующей потерей силы мышц в конечностях и в окрестностях туловища. В более чем половине случаев ходьба остается возможной до возраста 40 лет.
Миопатия Штейнтера. Это одна из трех наиболее распространенных миопатий у взрослых и чаще всего встречается в Квебеке. Симптомы варьируются от человека к человеку. Несмотря на то что они обычно появляются в возрасте 30−40 лет, существуют более ранние формы (ювенильные и врожденные).
Также наблюдается Миотония — аномальное и продолжительное сокращение мышц (мышца расслабляется слишком медленно), особенно выражается в руках, а иногда и на языке. Также могут быть затронуты мышцы лица, шеи и лодыжек. Часто присутствуют сердечные и дыхательные нарушения, которые являются потенциально серьезными. Нередко наблюдаются пищеварительные, гормональные, глазные расстройства, а также бесплодие и раннее облысение.
Миопатия поясничного отдела. Симптомы обычно проявляются в детстве (10 лет) или в раннем взрослом возрасте (около 20 лет). Мышцы плеч и бедер постепенно ослабевают, в то время как мышцы головы, шеи и диафрагмы обычно не затрагиваются. Если некоторые формы сопровождаются дыхательными нарушениями, то при этом типе дистрофии такие аномалии отсутствуют. Сердечные нарушения встречаются редко. Эволюция (развитие заболевания) очень изменчива, в зависимости от формы.
Миопатия Дежерина-Ландузи или плечелопаточная дистрофия. Симптомы обычно появляются в позднем детстве или в зрелом возрасте (от 10 до 40 лет). Как следует из названия, миопатия затрагивает мышцы лица, плеч и рук. Таким образом, больному становится сложно выразить улыбку, произнести некоторые предложения и закрыть глаза. Потеря подвижности происходит примерно в 20% случаев. Заболевание развивается медленно, продолжительность жизни нормальная.
Врожденные МД. Симптомы варьируются от одной формы к другой и присутствуют при рождении или в первые месяцы жизни. Ребенок имеет небольшой мышечный тонус, ему трудности сосать и глотать, иногда даже дышать. Эти дистрофии могут сопровождаться, в частности, пороками головного мозга, умственной отсталостью, аномальным развитием глаз.
Окуло-глоточная миотония. Это заболевание относительно распространено в Квебеке. Симптомы обычно появляются около 40 или 50 лет. Первые признаки болезни проявляются опустившимися веками, за которыми следуют слабость мышц глаз, лица и горла (глотки), вызывая трудности с глотанием пищи. Прогрессирование заболевания происходит медленно.
Исследования и прогресс
С 2005 года для лечения пациентов с развивающимся поражением мышц все чаще используются стволовые клетки. Для лечения мышечной дистрофии этим методом могут быть рассмотрены различные варианты заболевания, такие как: мышечные дистрофии Дюшенна, Беккера, миопатия поясничного и плечевого отдела.
Целью лечения является регенерация потерянных и поврежденных мышечных волокон с использованием регенеративного потенциала стволовых клеток. Для этого большое количество стволовых клеток вводится при помощи нескольких внутривенных и внутримышечных инъекций, что позволяет лучше нацеливать терапию именно на пораженную группу мышц.
Возможный прогресс
Терапия с применением стволовых клеток может обеспечить улучшение в плане мышечной массы, силы, движений, баланса, тремора и ригидности мышц. Стволовые клетки также могут замедлить будущую потерю мышечного объема и уменьшить симптомы.
Важно отметить, что лечение не является окончательным лекарством от этого заболевания и никоим образом не может решить проблему потери мышечных волокон. По этой причине прогресс после такого лечения не может быть постоянным. Исследования в этой области все еще ведутся.
Семейства заболевания
Обычно существуют два основных семейства МД:
- Мышечна врожденная дистрофия (ВМД), которая выражается в первые 6 месяцев жизни, сопровождает около десяти форм патологий переменной тяжести, включая ВМД с первичной недостаточностью мерозина, синдром Ульриха и Уокера-Варбурга;
- Мышечные дистрофии, появляющиеся в детстве или в зрелом возрасте:
![]()
Миопатия Дюшенна- Миопатия Беккера
- Миопатия Эмери-Дрейфуса (существует несколько форм)
- миопатия Ландузи-Дежерина
- Так называемая миопатия поясничного отдела — затрагивает мышцы вокруг плеч и бедер.
- Миотонические дистрофии (типы I и II), которые включают болезнь Штейнтера. Они характеризуются миотонией — когда мышцы не могут нормально расслабиться после сокращения.
- Окулофарингеальная миопатия
Эволюция дистрофии
Эволюция (развитие заболевания) МД сильно варьируется от одной формы к другой, а также от одного человека к другому. Некоторые формы быстро развиваются, что приводит к ранней утрате подвижности и ходьбе, а иногда и к смертельным сердечным или респираторным осложнениям, в то время как другие развиваются очень медленно — в течение десятилетий. Большинство врожденных мышечных дистрофий, например, которые мало выражены или почти незаметны, позже могут проявятся внезапно и с серьезными последствиями.
Возможные осложнения
Осложнения сильно различаются в зависимости от типа патологии. Некоторые нарушения могут затрагивать респираторные мышцы или сердце, иногда с очень тяжелыми последствиями.
Таким образом, сердечные осложнения довольно распространены, особенно у мальчиков с мышечной дистрофией Дюшенна.
Кроме того, дегенерация мышц заставляет тело и суставы деформироваться постепенно: на фоне этого у больных может развиваться сколиоз. Часто наблюдается сокращение мышц и сухожилий, что приводит к их стягиванию. Все эти нарушения приводят к деформации суставов: ноги и руки повернуты внутрь и вниз, деформируются колени или локти.
Также известно, что болезнь сопровождается тревожными или депрессивными расстройствами, поэтому больным требуется много внимания и поддержки, в первую очередь со стороны близких.
- Подписаться на AdMe
- Поделиться в Facebook
- Рассказать ВКонтакте
Ребята, мы вкладываем душу в AdMe.ru. Cпасибо за то,
что открываете эту красоту. Спасибо за вдохновение и мурашки.
Присоединяйтесь к нам в Facebook и ВКонтакте
Мышечная дистрофия обычно вызывается генетическими мутациями и приводит к прогрессирующему разрушению и потере мышечных клеток в организме. Она включает более 150 заболеваний, проявляющихся различным образом, но почти все они начинаются с едва заметных симптомов. Эти симптомы постепенно усугубляются, если вовремя не обратить на них внимание.
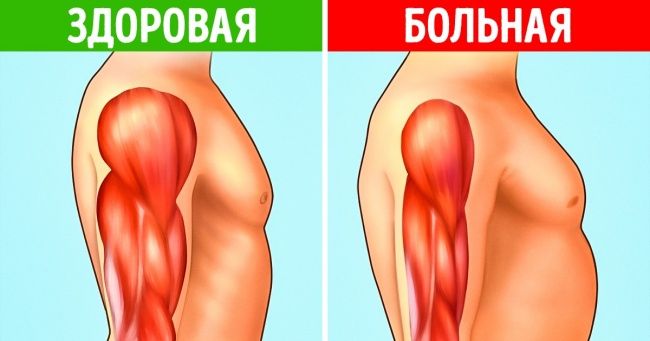
Потеря мышечных клеток обычно вызывает ощущение слабости в мышцах. Так что если вам тяжело вставать со стула, расчесывать волосы, поднимать предметы или вы постоянно что-то роняете, то, возможно, вы страдаете от мышечной дистрофии.
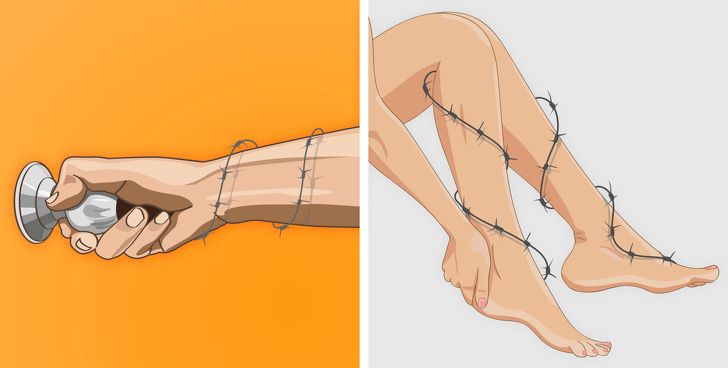
Вы также можете ощутить, что мышцы стянуты и утратили гибкость, а частая мышечная боль свидетельствует о том, что с ними явно что-то не в порядке. В то же время такие симптомы, как длительные спазмы, стянутость и напряжение в мышцах рук и ног, могут быть признаками миотонии — серьезного заболевания, требующего медицинского вмешательства.
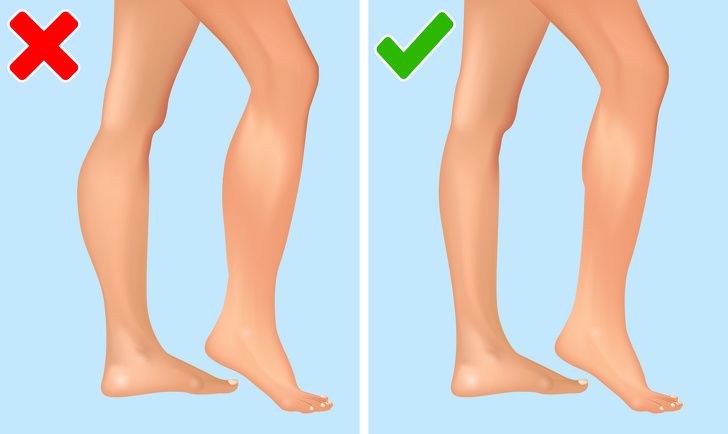
Увеличенные мышцы икр могут быть одним из признаков мышечной дистрофии Дюшенна. Эта болезнь обычно возникает у мальчиков в раннем возрасте и развивается очень быстро.
Если вас поразила болезнь Дюшенна, мышцы ваших икр страдают в первую очередь, так как на них идет большая нагрузка, связанная со стабилизацией всего тела. При этом мышцы постепенно заменяются жиром и рубцовой тканью.
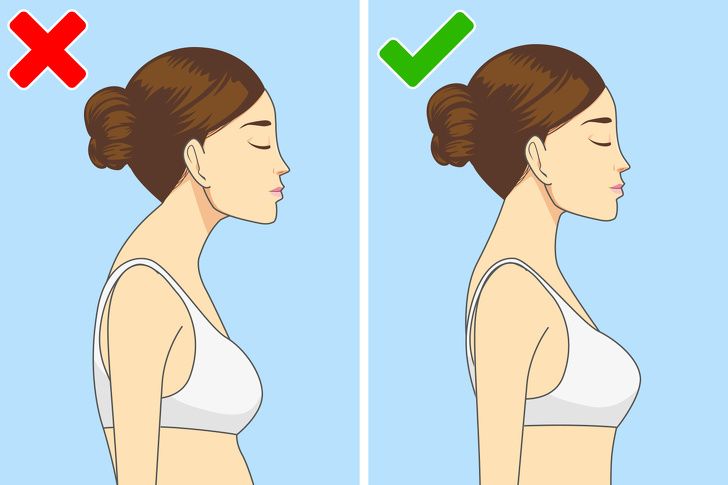
Если у вас недостаточно сильные мышцы, чтобы держать спину ровно, у вас может быть плохая осанка, которая со временем приведет к сколиозу. При этом заболевании ваша спина искривляется в правую или левую сторону, что приводит к смещению внутренних органов.
Сколиоз обычно проявляется в подростковом возрасте и чаще диагностируется у женщин, чем у мужчин. У этой болезни есть множество негативных последствий для здоровья, включая головные боли и постоянную боль в ногах.

В тяжелых случаях сколиоза прогрессирующая мышечная слабость может влиять на мышцы груди, связанные с процессом дыхания. Хотя вы можете не испытывать трудностей с дыханием как таковым, но вы можете страдать от проблем, указывающих на плохую работу респираторной системы, среди которых головные боли, невозможность сконцентрироваться и ночные кошмары.
Слабые мышцы груди затрудняют кашель, увеличивая риск серьезных инфекций дыхательных путей. Если обычную простуду не лечить как следует, она может быстро перерасти в пневмонию.

Проблемы с едой, включающие потерю способности жевать и проглатывать еду, кашель, першение или глухой голос после еды, могут быть признаками болезни Кеннеди. Это заболевание может проявиться в любом возрасте, но в большинстве случаев развивается в среднем взрослом возрасте.
Наряду с трудностями с едой, вы можете столкнуться с другими симптомами типа изменения речи, гнусавости и даже полной атрофии мышц лица, челюсти и языка. Все они требуют немедленного медицинского лечения.

Некоторые формы мышечной дистрофии могут вызывать прогрессирующие изменения в работе сердца. Эти изменения называются кардиомиопатией и на ранней стадии могут проходить бессимптомно, хотя некоторые старадают от одышки, усталости и отека ног.
Однако из-за этой болезни мышцы сердца не могут качественно работать. Поэтому со временем могут появиться симптомы типа нерегулярного сердцебиения, обмороков и головокружения.

В дополнение к истощению мышечной ткани, люди с миотонической дистрофией часто страдают от различных глазных болезней. К этим проблемам относятся слабость глазных мышц, слезящиеся глаза, низкое глазное давление и катаракта.
Катаракта — самый типичный признак мышечной дистрофии. Для нее характерно затуманивание хрусталика глаза, что обычно становится причиной размытого зрения, тусклых цветов, проблем с восприятием яркого света и ночным зрением.

Некоторые мужчины, страдающие от миотонической дистрофии, также сталкиваются с гормональными изменениями. Эти гормональные нарушения обычно провоцируют раннее облысение передней части головы, обычно происходящее в возрасте между 20 и 30 годами.
Эндокринное нарушение также может привести к импотенции и атрофии яичек. Как правило, эта болезнь вызывает бесплодие.

Хотя лечения как такового не существует, вы все же можете попробовать выполнять простые действия, чтобы поддерживать мышцы сильными и здоровыми.
- Упражняйтесь регулярно. Так как мышцы постепенно слабеют, один из лучших способов замедлить этот процесс - обеспечить ежедневную физическую нагрузку. Регулярные тренировки с низкой интенсивностью и растяжки помогут вам стимулировать тело и естественным путем нарастить мышечную массу.
- Ешьте больше продуктов, богатых витаминами Е и D. Если вы страдаете от слабости в мышцах, потребляйте больше лосося, сардин, креветок, сыра, яиц, миндаля, авокадо, брокколи и оливкового масла.
- Используйте куркуму во время готовки. Эта старинная индийская приправа содержит мощное вещество, куркумин, которое весьма полезно для предотвращения и лечения мышечного истощения.
- Пейте зеленый чай вместо других напитков. Согласно некоторым исследованиям, около 7 чашек зеленого чая в день могут замедлить износ мышечной ткани, борясь с оксидативным стрессом в мышцах.
- Добавляйте пищевую соду в ванну. Благодаря своей щелочной природе это вещество защищает ваши мышцы и снимает боль и воспаление в них.
Если вы заметили один из этих симптомов, первым же делом обратитесь к врачу.
Знаете другие способы лечения мышечной дистрофии? Поделитесь своими знаниями по этой теме в комментариях ниже. Будьте здоровы!
Медицинский справочник болезней
Миопатия. Формы, причины, симптомы и лечение миопатий.
Миопатия — прогрессирующая мышечная дистрофия — сборная группа заболеваний, характеризующихся первичным дистрофическим процессом в мышечной ткани.
Причины.
Относится к наиболее часто встречающимся хроническим заболеваниям нервно-мышечного аппарата и носит наследственный характер.
Различные экзогенные вредности (травмы, инфекция, интоксикация) могут выявить имеющуюся патологию или вызвать ухудшение текущего процесса. Для установления семейного характера заболевания необходим не только тщательный анамнез, но и по возможности более полный осмотр всех членов семьи с выявлением так называемых малых признаков заболевания.
Наличие спорадических случаев не исключает наследственную природу.
Следует также иметь в виду возможность фенокопий миопатии, т.е. симптоматических форм или миопатических синдромов.
Патологоанатомия.
При патологоанатомических исследованиях в нервной системе не находят характерных изменений. В редких случаях отмечаются незначительное уменьшение клеток передних рогов спинного мозга, иногда — изменения в двигательных нервных окончаниях в виде набухания миелиновой оболочки, изменения осевых цилиндров. В моторных бляшках исчезает фибриллярная структура. Грубые изменения находят в поперечнополосатых мышцах. Мышцы истончены, большая часть волокон замещена соединительной тканью и жиром. Характерна неравномерность отдельных мышечных волокон — одни волокна резко уменьшены, другие, наоборот, резко увеличены.
В поздних стадиях заболевания почти вся мышечная ткань замещена соединительной или жировой тканью. В сосудах наблюдается пролиферация адвёнтиции, сужение просвета и иногда пристеночное тромбообразование. По мере развития процесса увеличивается общая масса эндо- и перимизиальной соединительной ткани с образованием фиброзного футляра вокруг мышечных волокон и внутримышечных кровеносных сосудов.
При гистохимическом исследовании наблюдается увеличение кислых муко- полисахаридов в основном веществе мышц и коллагеновых волокнах.
Патогенез.
Патогенез миопатий до настоящего времени неясен. Первичный биохимический дефект не установлен. Наиболее значительным изменениям подвергается белковый и углеводный обмен в мышечной ткани. В последнее время высказана гипотеза о нарушении обмена циклических нуклеотидов (циклических АМФ и ГМФ), являющихся универсальными регуляторами внутриклеточного обмена и ответственными за реализацию генетической информации.
Клиническая картина.
Симптоматика миопатий характеризуется нарастающими атрофиями произвольной мускулатуры. Параллельно развитию мышечного похудания появляются и парезы, однако мышечная слабость обычно выражена меньше, чем степень атрофии.
В связи с медленным прогрессированием процесса и неравномерностью поражения отдельных мышечных групп и даже участков мышц создаются условия для относительной компенсации двигательного дефекта: больные миопатией длительное время остаются трудоспособными и могут себя обслуживать, прибегая к ряду характерных вспомогательных движений. Постепенно угасают сухожильные рефлексы. Чувствительность, координация движений не нарушаются. Тазовые функции всегда сохранены.
Для некоторых видов миопатий характерны наличие псевдогипертрофий, наклонность к концевым атрофиям и сухожильным ретракциям. Фасцикулярные подергивания отсутствуют. Механическая возбудимость мышц снижена.
Нередко наблюдаются те или иные изменения внутренных органов,
- главным образом сердца: расширение границ, глухость тонов, нарушение проводимости, подтверждаемое электрокардиограммой;
- страдает функция внешнего дыхания;
- вегетативные нарушения выражаются цианозом кистей и стоп, резко повышенной потливостью, похолоданием дистальных отделов конечностей, асимметрией кожной температуры, пульса, повышенным пиломоторным рефлексом;
- страдает микроциркуляция в мышцах конечностей;
- рентгенография трубчатых костей позволяет обнаружить дистрофические явлени я;
- при электромиографическом исследовании констатируется характерная картина — снижение амплитуды биопотенциалов при достаточной частоте, а также укорочение длительности одиночного потенциала и полифазный характер;
- при биохимических исследованиях находят нарушения в креатин-креатининовом обмене. Почти всегда в моче значительно уменьшается количество креатинина и появляется креатин. Креатиновый показатель в известной степени говорит о тяжести дистрофического процесса. Отмечается значительная аминоацидурия. При ряде форм миопатий очень рано (в преклинической или в начальной клинической стадии) можно обнаружить увеличение ферментов в сыворотке крови. В первую очередь это касается специфического для мышечной ткани фермента креатинфосфокиназы;
- увеличивается также содержание аминофераз и альдолаз . Уменьшена артериовенозная разница содержания сахара крови, увеличено содержание пировиноградной и молочной кислот в мышцах и в крови. Как правило, уменьшен уровень лимонной кислоты в крови. Имеются данные о специфичности некоторых биохимических изменений при различных типах миопатий.
Классификация миопатий.
До настоящего времени нет достаточно обоснованной и общепринятой классификации миопатий. В большинстве случаев используют классификацию, основанную на клиническом принципе.
Псевдогипертрофическая форма Дюшенна является одной из наиболее частых форм миопатий. Она характеризуется самым ранним началом заболевания — нередко с 2—5-летнего возраста, а иногда даже с первого года жизни, и наиболее злокачественным течением. В типичных случаях дети к 10—12 годам уже с трудом ходят и к 15 годам становятся полностью обездвиженными.
В первую очередь страдают мышцы проксимальных отделов нижних конечностей, тазового пояса, затем в процесс вовлекаются мышцы проксимальных отделов рук. Рано выпадают коленные рефлексы. Характерна псевдогипертрофия икроножных мышц; часто уплотнение и гипертрофия их являются первым симптомом заболевания.
Псевдогипертрофии могут наблюдаться и в других группах мышц — ягодичных, дельтовидных, иногда - в языке; отмечаются значительно выраженные ретракции, в первую очередь со стороны ахилловых сухожилий. Нередко страдает сердечная мышца. Отмечается снижение интеллекта в разной степени выраженности. Для миопатии Дюшенна очень характерен высокий уровень ферментов сыворотки крови, особенно креатинфосфокиназы. Повышенный уровень ферментов можно обнаружить и у носительниц мутантного гена.
Заболевание передается по рецессивному, сцепленному с Х-хромосомой типу. Болеют только мальчики, матери являются кондукторами. Риск заболевания сыновей матерей-носительниц 50 %; 50 % дочерей становятся носителями патологического гена. Пенетрантность высокая.
Доброкачественная псевдогипертрофическая миопатия, сцепленная с Х-хромосомой (миопатия Беккера), выделена в самостоятельную форму в связи с рядом особенностей. Начало заболевания — чаще между 12 и 25 годами, иногда раньше. Течение мягкое, прогрессирование медленное, больные многие годы сохраняют трудоспособность или самообслуживание. Интеллект всегда сохранен. В остальном клиническая картина подобна псевдогипертрофической форме Дюшенна.
Плечелопаточно-лицевая форма Ландузи — Дежерина — относительно часто встречающийся тип миопатии.
Начинается заболевание, как правило, в детском или юношеском возрасте; течение сравнительно благоприятное. Первые симптомы касаются поражения мышц лица, особенно круговой мышцы рта, или мышц плечевого пояса. К этому присоединяется слабость и похудание мышц проксимальных отделов рук, затем развивается парез дистальных отделов ног. Наблюдаются умеренные гипертрофии мышц, а затем своеобразные патологические позы из-за неравномерности атрофии различных групп мышц и ретракций. Сухожильные рефлексы долго остаются сохраненными. Может быть асимметрия поражения.
Заболевание передается по аутосомно-доминантному типу с полной пенетрантностью, страдают в равной степени мужчины и женщины. Отмечаются отчетливые различия в тяжести клинических признаков не только в разных семьях, но и у разных членов одной семьи (могут быть тяжелые, легкие и абортивные формы).
Описаны стертые формы. В сыворотке крови находят умеренное повышение ферментов. Начало заболевания очень вариабельно— от детского возраста до сравнительно зрелого, но чаще в начале второго десятилетия, что отражает название этой формы. Также изменчив характер течения — иногда мягкое, благоприятное, иногда очень злокачественное.
Большинство авторов признают аутосомно-рецессивный тип наследования заболевания. Часты спорадические формы и фенокопии. Болеют одинаково часто мужчины и женщины.
Дистальная форма миопатии встречается редко.
Характеризуется поражением мышц голеней, стоп, предплечий, кистей, постепенно процесс генерализуется. Отмечаются ретракции, концевые атрофии мышц. Заболевание начинается в сравнительно позднем возрасте — 20—25 лет; прогрессирование, как правило, медленное. Отсутствие расстройств чувствительности, нормальная скорость проведения возбуждения, повышенный уровень сывороточных ферментов отличают дистальную миопатию от невральной амиотрофии.
Тип наследственной передачи—аутосомно-доминантный с неполной пенетрантностью. Несколько чаще болеют лица мужского пола.
Лопаточно-перонеальная амиотрофия (миопатия Давиденкова) проявляется поражением дистальных мышц нижних и проксимальных отделов верхних конечностей и мышц плечевого пояса. Начинается заболевание сравнительно поздно — в 25—30 лет. Наблюдаются концевые атрофии, например в большой грудной мышце, иногда крупные фасцикулярные подергивания, раннее угнетение сухожильных рефлексов.
В ряде случаев отмечаются легкие расстройства чувствительности — дистальные парестезии, гипестезии, иногда умеренная боль. Почти никогда не бывает креатинурии.
При электромиографическом исследовании выявляются специфические изменения, отличающие эту форму от обычной миопатии и невральной амиотрофии Шарко — Мари (дизритмичные колебания в покое, снижение амплитуды, уменьшение частоты и иногда групповые спайковые разряды при активных движениях).
Таким образом, данная форма является как бы промежуточной между первичной миопатией и невральной амиотрофией. Некоторые авторы рассматривают эту форму лишь как особый вариант (скапуло-перонеальный синдром) плечелопаточно-лицевой миопатии Ландузи — Дежерина.
Редкие варианты миопатии.
Описано большое количество различных вариантов прогрессирующего мышечного поражения наследственного характера. Так, например, описаны миопатия четырехглавой мышцы бедра, миосклеротическая миопатия, мышечная дистрофия с истинными гипертрофиями, врожденная мышечная дистрофия с медленным и быстрым прогрессированием, иногда с катарактой, мышечный инфантилизм и др.
Непрогрессирующие миопатии включают группу заболеваний, отличающихся или своеобразными изменениями строения мышечных клеток, или специфическими биохимическими нарушениями. Проявляются эти состояния сравнительно рано, обычно на 1—3-м году жизни, имеют сравнительно благоприятное течение. Диагноз может быть поставлен после биопсии мышц, иногда только после электронно- микроскопического исследования.
Болезнь центрального стержня характеризуется резким снижением или полным отсутствием ферментативной активности в центральной части мышечного волокна, что выявляется при окраске препарата мышечной ткани трехвалентным хромом по Гомори.
Клиническая картина: снижение мышечного тонуса, дряблость мышц, задержка развития двигательных функций, в позднем возрасте — умеренная слабость проксимальных отделов и гипотрофия мышц.
На ЭМГ — уменьшение длительности колебаний потенциала и увеличение полифазных потенциалов. Передача по доминантному типу с неполной пенетрантностью. Часты спорадические случаи.
Немалиновая, или нитеобразная, миопатия проявляется врожденной мышечной слабостью конечностей и лица с понижением мышечного тонуса и отсутствием сухожильных рефлексов. Описаны изменения скелета, в частности деформация грудной клетки, сколиоз.
На ЭМГ — изменения, характерные для мышечного уровня поражения. При электронно-микроскопическом исследовании выявляются своеобразные нитевидные структуры под сарколеммой.
Миотувулярная миопатия клинически выражается в понижении мышечного тонуса, умеренно выраженных атрофиях мышц конечностей с наличием диффузной слабости рук, ног, туловища. Характерны также слабость мимической мускулатуры, птоз и ограничение подвижности глазных яблок, общая задержка развития двигательных функций. Состояние может быть стационарным или медленно прогрессирующим. У большинства больных выявляются те или иные костные деформации.
На ЭМГ — сочетание мышечного типа изменений с наличием спонтанной активности. Гистологически определяются мышечные волокна резко уменьшенной величины с центральным расположением ядер, напоминающие по строению эмбриональную мышечную ткань. При электронной микроскопии выявляются участки дегенеративно измененных миофибрилл, при гистохимическом исследовании обнаруживается повышение активности митохондри- альных ферментов.
Митохондриальные миопатии характеризуются увеличением числа митохондрий в мышечных волокнах или увеличением размеров митохондрий, обнаруживаемых при электронно-микроскопическом исследовании. Клинически отмечается мышечная слабость, главным образом в проксимальных отделах рук и ног, вялость, быстрая утомляемость при отсутствии особых мышечных атрофий. Прогрессирования, как правило, не отмечается.
Диагноз прогрессирующей мышечной дистрофии, как правило, не представляет больших трудностей. Атипичные формы приходится дифференцировать от сирингомиелии (переднероговая форма), начальных явлений амиотрофического склероза, спинальных амиотрофий, полимиозита и других миопатических синдромов (см.). Комплексное обследование больного с применением биохимических (определение уровня ферментов и др.), электрофизиологических (ЭМГ, определение скорости распространения возбуждения по нерву), гистологических исследований и анализ клинической картины позволяют поставить правильный диагноз.
ЛЕЧЕНИЕ.
- Воздействие на энергетической обмен в мышцах осуществляется назначением АТФ в виде монокальциевой соли по 3—6 мл в день внутримышечно в течение 30 дней.
- Показано применение витамина Е внутрь по 30—40 капель 3 раза в день или внутримышечно раствор токоферола ацетата в масле по 1—2 мл — 20 инъекций (или эревит).
- Назначают витамины группы В, гликокол, лейцин (по 1 столовой ложке 3 раза в день), глутаминовую кислоту (по 0,5—1 г 3 раза в день).
Накопленный опыт по применению анаболических гормонов не подтверждает возлагавшихся на этот вид лечения надежд.
- Рекомендуется сочетать медикаментозное лечение с физиотерапией (гальванический воротник и гальванические трусы с кальцием, солянохвойные ванны со строго индивидуальной лечебной физкультурой при средней нагрузке).
- При наличии контрактур может быть рекомендовано оперативное вмешательство на сухожилиях.
- Показан систематически проводимый легкий массаж.
Читайте также:
