
Что за болезнь когда кости неправильно растут
Что такое оссифицирующая прогрессирующая фибродисплазия?
Оссифицирующая прогрессирующая фибродисплазия (или сокр. ОПФ, болезнь Мюнхеймера, синдром каменного человека) — очень редкое наследственное заболевание, при котором соединительные ткани тела, включая мышцы, сухожилия и связки, постепенно замещаются костью (в процессе, называемом оссификацией).
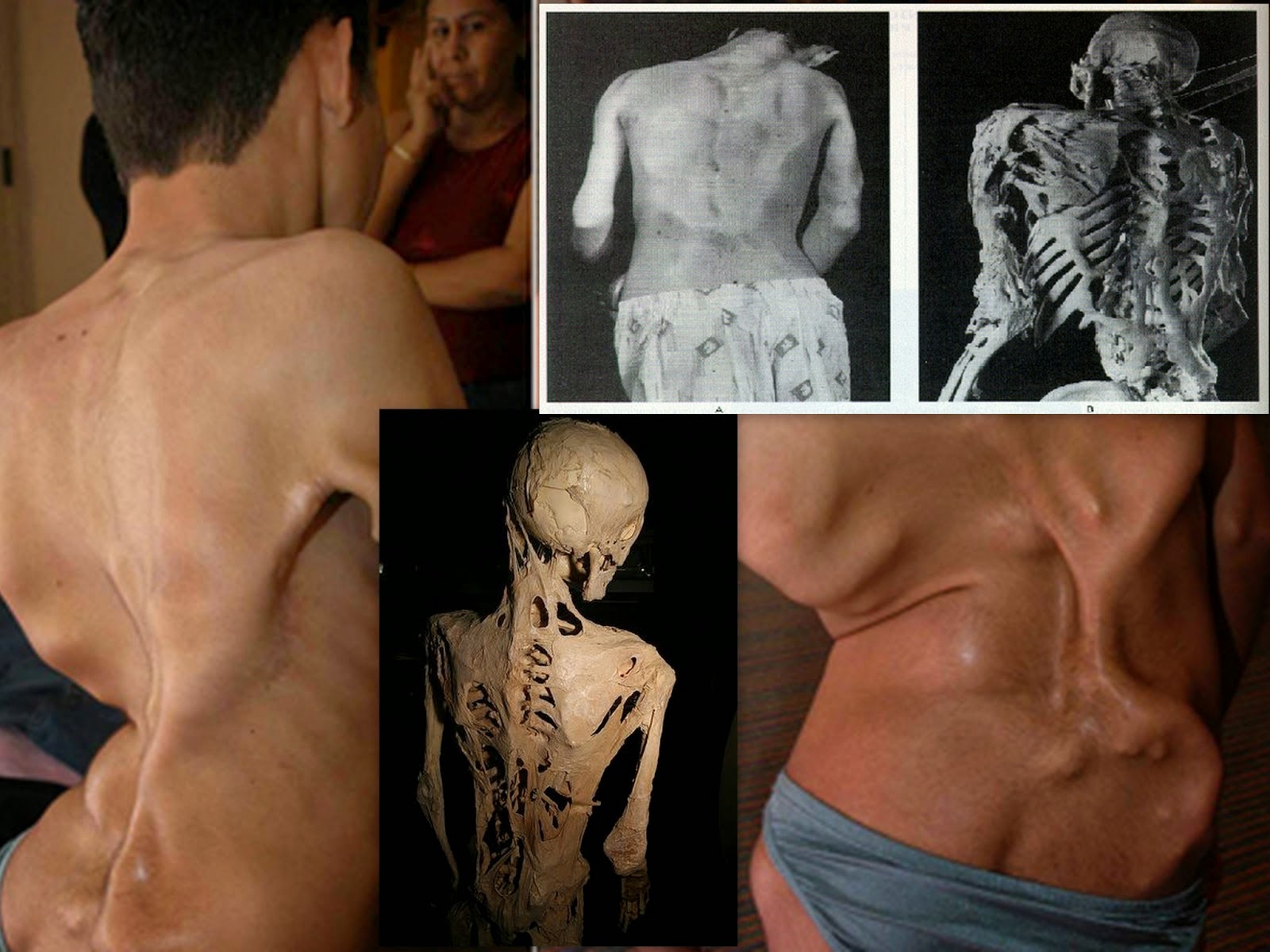
Болезнь Мюнхеймера присутствует при рождении, однако симптомы могут не проявляться до раннего детства. Окостенение может произойти случайно или после травмы.
Симптомы ОПФ
У рожденных с ОПФ, признаки и симптомы окостенения могут никак не выявляться, пока ребенок не станет немного старше и не начнет расти.
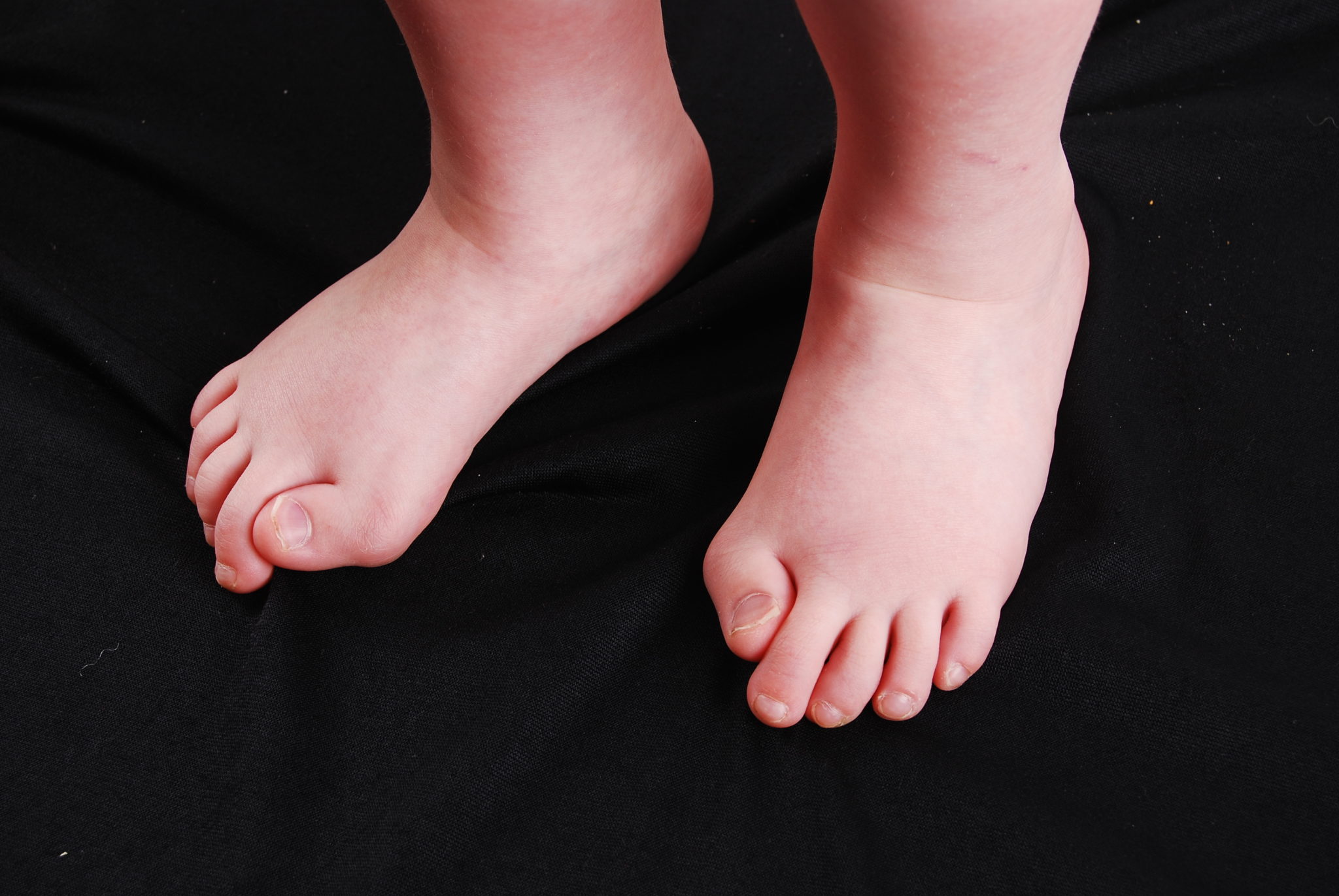
У новорожденных первым признаком ОПФ часто является врожденная аномалия пальцев ног.
Вскоре после рождения медицинские работники или родители могут заметить, что большие пальцы ноги ребенка короче других пальцев (см. фото выше).
Этот порок развития наблюдается у всех людей с синдромом каменного человека и является важной подсказкой для постановки диагноза.
У новорожденного также могут быть отеки вокруг глаз и кожи головы. В некоторых случаях этот отек может начаться, когда плод еще находится в утробе матери, хотя это состояние обычно диагностируется только после рождения. Около 50 процентов людей с этим заболеванием также имеют сходные врожденные аномалии в больших пальцах — также наблюдались и другие пороки развития, например, в позвоночнике.
В то время как общая скорость прогрессирования состояния неизвестна, окостенение имеет тенденцию следовать определенной схеме, начиная с шеи и вплоть до плеч, туловища, конечностей и ступней.
Однако, поскольку на формирование кости могут повлиять травма (например, перелом руки) или вирусное заболевание (например, грипп), заболевание может не строго следовать этому прогрессу.
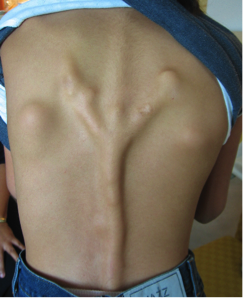
Основные симптомы болезни Мюнхеймера зависят от того, какие части тела стали окостеневшими. Часто, при заболевании встречаются нежные комки под кожей (подкожные узелки, см. фото выше). Иногда формированию этих узелков предшествует умеренная лихорадка. Большинство людей с ОПФ будут иметь общие симптомы боли, ригидности и прогрессирующего недостатка подвижности по мере разрастания костной ткани.
В зависимости от того, какие части тела окостенеют, более специфические симптомы ОПФ могут включать:
- проблемы с питанием, которые могут привести к дефициту питательных веществ или недоеданию;
- трудно с разговором;
- проблемы с зубами;
- затрудненное дыхание;
- респираторные инфекции;
- нарушение слуха;
- выпадение волос (облысение);
- анемию;
- сдавливание нервов;
- правосторонняя застойная сердечная недостаточность;
- искривление позвоночника (сколиоз и кифоз).
- сенсорные аномалии;
- умеренную умственную отсталость;
- неврологические симптомы.
Люди с синдромом каменного человека могут иметь периоды в своей жизни, когда костная ткань не будет расти. В других случаях может показаться, что это явление происходит случайно и при отсутствии каких-либо очевидных травм или заболеваний. Если оссификация происходит в необычной части тела (где кости обычно не обнаруживаются), это может привести к переломам.
Со временем формирование новой опухоли костей и тканей, которая сопровождает это состояние, может серьезно повлиять на то, насколько хорошо человек может двигаться.
В большинстве случаев оссифицирующая прогрессирующая фибродисплазия в конечном итоге приводит к полной иммобилизации. Многие люди с этим заболеванием к 30 годам будут прикованными к постели.
Причины фибродисплазии
У большинства людей, рожденных с ОПФ, заболевание возникает в результате случайной генетической мутации. Хотя это генетическое заболевание, оно обычно не встречается у всей семьи.
Человеку нужен только один пораженный ген, чтобы у него возникла оссифицирующая прогрессирующая фибродисплазия. Большинство случаев происходит из-за случайной мутации — у человека редко развивается состояние, потому что только от одного из родителя наследуется ненормальный ген. В генетике это явление называется как аутосомно-доминантное расстройство.
Мутация гена, ответственная за состояние, была идентифицирована исследователями из Университета Пенсильвании — они идентифицировали рецептор на хромосоме 2, называемый рецептором активина типа IA (ACVR1). ACVR1 присутствует в гене, который кодирует костные морфогенетические белки (BMP), которые помогают формировать и восстанавливать скелет, начиная с момента образования эмбриона.
Исследователи полагают, что мутация в гене предотвращает отключение этих рецепторов, что позволяет неконтролируемой кости образовываться в тех частях тела, где она обычно не появляется в течение жизни человека.
Диагностика
Оссифицирующая прогрессирующая фибродисплазия встречается очень редко. Предполагается, что только несколько тысяч человек страдают этим заболеванием, и в мире существует только около 800 известных пациентов с этим заболеванием — 285 из них находятся в Соединенных Штатах. По-видимому, ОПФ чаще встречается у детей определенной расы, и такое состояние встречается у мальчиков так же часто, как и у девочек.
Диагностика оссифицирующей прогрессирующей фибродисплазии может быть затруднена. Нередко изначально это заболевание ошибочно диагностируется как форма рака или как состояние, называемое агрессивным ювенальным фиброматозом.
В начале диагностики болезни Мюнхеймера, если ткань подвергается биопсии и исследованию под микроскопом (гистологическое исследование), она может иметь некоторые сходства с агрессивным ювенильным фиброматозом. Однако при последнем заболевании поражения не прогрессируют до полностью сформированной кости, как при синдроме каменного человека. Это может помочь врачу провести различие между данными болезнями.
Одним из основных диагностических признаков, который может привести врача к подозрению на болезнь Мюнхеймера, в отличие от другого состояния, является наличие коротких, неправильно сформированных больших пальцев ног.
Если биопсия ткани неясна, клиническое обследование ребенка может помочь врачу исключить агрессивный ювенильный фиброматоз. У детей с агрессивным ювенильным фиброматозом нет врожденных пороков развития пальцев ног или рук, однако она почти всегда встречается у детей с ОПФ.
Другое состояние, прогрессирующая костная гетероплазия, может также быть перепутана с оссифицирующей прогрессирующей фибродисплазией. Основное различие при постановке диагноза заключается в том, что рост кости при прогрессирующей костной гетероплазии обычно начинается на коже, а не под ней. Эти костные бляшки на поверхности кожи отличаются от нежных узелков, которые образуются при ОПФ.
Другие методы обследования, которые доктор может назначить, если есть подозрения на синдром каменного человека, включают:
- полный медицинский анамнез и медицинский осмотр;
- рентгенологические исследования, такие как компьютерная томография (КТ) или сцинтиграфия костей (сканирование костей) для поиска изменений скелета;
- лабораторные анализы для измерения уровня щелочной фосфатазы;
- генетическое тестирование для поиска мутаций.
Если подозревается наличие ОПФ, врачи стараются избегать любых инвазивных тестов, процедур или биопсии, потому что травма обычно приводит к образованию большего количества костной ткани у человека с таким заболеванием.
Хотя состояние обычно не встречается у всей семьи, родителям, у которых есть ребенок с диагнозом болезни Мюнхеймера, может быть полезна медико-генетическая консультация.
Лечение
В настоящее время нет лекарства от ОПФ. Также нет определенного или стандартного курса лечения. Существующие методы терапии не эффективны для каждого пациента, и поэтому основной целью является лечение симптомов и предотвращение роста костей, если это возможно.
Хотя терапия не остановит прогрессирование состояния, медицинские решения по лечению боли и других симптомов, связанных с ОПФ, будут зависеть от потребностей конкретного пациента. Врач может порекомендовать попробовать один или несколько из следующих способов лечения препаратами и процедурами, чтобы улучшить качество жизни пациента:
- Преднизон в высоких дозах или другой Кортикостероид;
- такие препараты, как Ритуксимаб (обычно используется для лечения ревматоидного артрита);
- Ионтофорез лекарственных веществ для доставки лекарств через кожу;
- Миорелаксанты или инъекции местного анестетика;
- препараты под названием Бисфосфонаты, которые используются для защиты плотности костей;
- Нестероидные противовоспалительные препараты (НПВП);
- лекарства для подавления тучных клеток, которые могут помочь уменьшить воспаление.
Окостенение часто происходит случайным образом и не может быть полностью предотвращено, однако, в редких случаях, оно также может происходить в ответ на воспаление, травму и болезнь.
Таким образом, рекомендации относительно активности, образа жизни, профилактического ухода и вмешательств могут быть назначены начиная с детства.
Рекомендации могут включать:
- избегать ситуаций, которые могут привести к травме, например, полное исключение занятий спортом;
- избегать инвазивных медицинских процедур, таких как биопсия, стоматологических процедур и внутримышечной иммунизации;
- профилактика антибиотиками для защиты от болезней или инфекций, когда это необходимо;
- меры по профилактики инфекций, например, надлежащая гигиена рук, для защиты от распространенных вирусных заболеваний (таких как грипп) и других респираторных вирусов, а также от осложнений, таких как пневмония;
- физиотерапия;
- средства передвижения и другие вспомогательные устройства, такие как ходунки или инвалидная коляска;
- другие медицинские устройства, которые могут помочь в повседневной жизни для переодевания и купания.
- медицинские устройства или другие меры безопасности для предотвращения падений, например, при вставании с постели или приеме душа;
- психологическая и социальная поддержка пациентов и их семей;
- образовательная поддержка, включая специальное образование и обучение на дому;
- для семей может быть полезным медико-генетическая консультация.
Инвазивные процедуры или операции, направленные на удаление областей аномального роста кости, не рекомендуются, так как травма от операции почти всегда приводит к развитию дальнейшей окостенения.
Если операция абсолютно необходима, следует использовать наиболее минимально инвазивную методику. Пациентам с ОПФ также могут потребоваться особые анестезирующие средства.
В последние годы было проведено несколько клинических испытаний с целью разработки лучших вариантов лечения для людей с фибродисплазией.
Резюмируя
Оссифицирующая прогрессирующая фибродисплазия является чрезвычайно редким состоянием, при котором мутация гена заставляет соединительные ткани тела, включая мышцы, сухожилия и связки, постепенно замещаться костью (процесс называемый окостенением).
От болезни Мюнхеймера не существует лекарств и диагностика состояния довольно сложна.
Лечение в основном поддерживающее, и прогрессирование состояния обычно довольно непредсказуемо. Принятие мер во избежание травм и других ситуаций, которые могут привести к усилению окостенения, может помочь уменьшить количество замещений у человека, однако новая кость может все еще образовываться без какой-либо явной причины.
ОПФ обычно приводит к полной неподвижности, и к 30 годам большинство людей приковано к постели. В настоящее время проводятся клинические испытания, которые, как мы надеемся, позволят найти лучшие варианты лечения для повышения качества жизни пациентов с этим заболеванием.
Видеозаписи по теме
Генетические заболевания обусловлены патологическими нарушениями строения генома. "Дефектный" ген может быть получен от одного из родителей и проявиться как на 100%, так и на 10%. А вот болезни с наследственной предрасположенностью значительно отличаются от генетических. Если последние излечить невозможно, то заболевания, к которым человек имеет наследственную предрасположенность, возможно нивелировать рациональным питанием, здоровым образом жизни и профилактическими мерами.
Пять генетический заболеваний позвоночника и костей
Такие болезни напрямую связанны с нарушениями генома и проявляются в виде дефектов развития скелета человека. Генетические заболевания обусловлены нерациональным формообразованием ткани или нарушениями роста. Подобные болезни носят в медицине общие название - дисплазии.
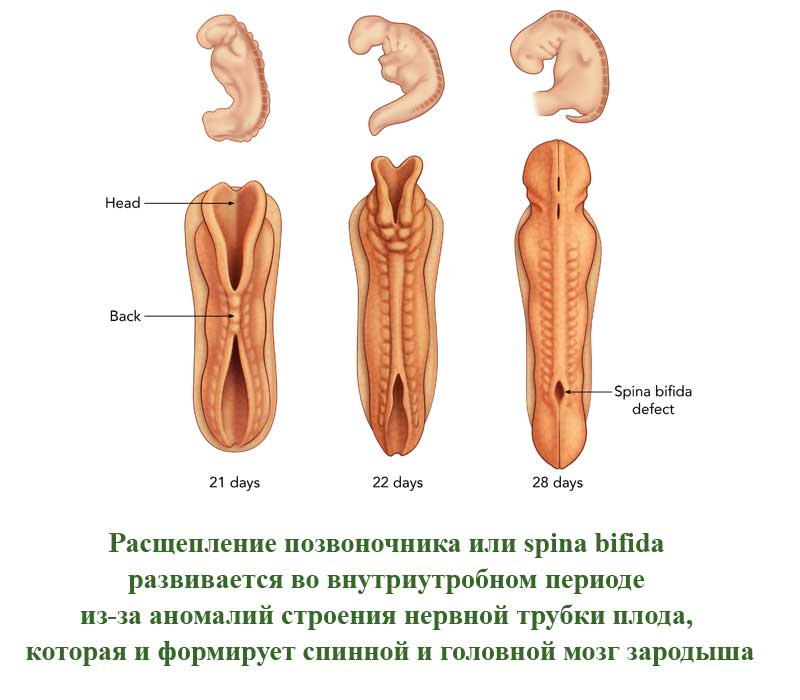
Это порок развития позвоночного столба, которое проявляется в виде недоразвитых позвонков. Такие позвонки не сомкнуты, через щель может быть виден спинной мозг. Заболевание развивается во внутриутробном периоде из-за аномалий строения нервной трубки плода, которая и формирует спинной и головной мозг зародыша. Расщепление позвоночного столба может проявляться и в закрытом виде, когда спинной мозг не виден снаружи.
В легких случаях заболевание могут обнаружить лишь при рентгеновском обследовании. А вот при самых серьезных формах болезни у ребенка могут сразу же образовываться свищи в полости позвоночника. Очень часто заболевание в тяжелых формах сопровождается параличом нижней части тела.
В более, чем 80% случаев, расщепление позвоночника сопровождается гидроцефалией спинного мозга и пороками развития головного мозга, а также - черепа.
По американской статистике, заболевание встречается у одного пациента из 1500. Российская статистика приводит следующие данные - 3 случая на 10000 человек. Однако, многие случаи расщепления позвоночника на территории СНГ остаются нераспознанными у новорожденных из-за легкой формы болезни.
Болезнь часто именуют остеопетрозом. Может протекать в двух формах:
- замедленной;
- злокачественной.
Генетическое заболевание встречается с частотой в 1 случай на 20000 пациентов. Для остеопетроза характерны такие симптомы:
- повышенная ломкость костей;
- увеличение плотности костной ткани;
- уменьшение размеров костномозговых лакун;
- нарушение гемопоэза;
- уменьшение массы костного мозга.
Генерализированный остеоклероз проявляется в достаточно раннем возрасте в виде разных беспорядочных слоев клеток костной ткани, увеличения общей массы костей и замедленном росте скелета.
При злокачественном течении болезни часто возникают внезапные переломы костей, развивается геморрагичекий синдром, жировая дистрофия органов, нарушается дентиногеез. Характерен очень небольшой рост.
В случае замедленного остеопетреза болезнь может быть выявлена лишь в 50% и протекать абсолютно бессимптомно. Выявляют заболевание случайно во время рентгена. В некоторых случаях может наблюдаться симптоматика синдрома "Кость внутри кости".
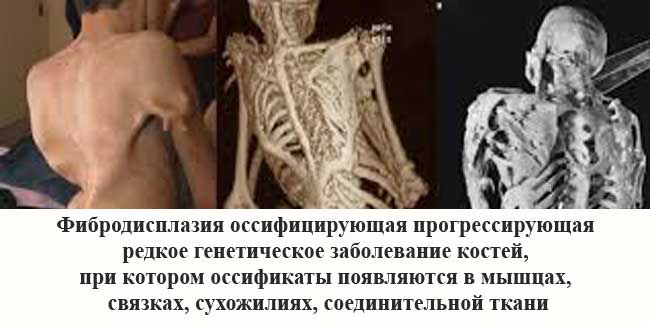
ФОП - это генетическое и очень редкое заболевание костей. При такой болезни организм начинает формировать новую костную массу в виде оссификатов в ненадлежащих местах тела, а именно внутри:
- соединительных тканей;
- связок;
- мышц;
- сухожилий.
К образованию оссификатов в организме может привести абсолютно любая травма: порез, операция, ушиб, внутримышечная инъекция или перелом. Поэтому образования такого типа удалять нельзя - на их месте костная ткань разрастется еще больше. По физиологическим признакам оссификаты совершенно не отличаются от здоровых костей.
Проблема лишь в неправильном расположении образования костной ткани. Возникает ФОП из-за мутаций гена ACVR1/ALK2. Данный ген кодирует рецептов костного морфогенетического белка. Носителем гена быть невозможно, его наличие в теле всегда вызывает развитие фибродисплазии оссифицирующей. Передается заболевание по наследству и на данный момент является неизлечимым.
Такие заболевания характеризуются чрезмерным развитием костной массы. Носят общее название - остеохондродисплазии. Гиперостозы возникают из-за генетических нарушений и патологий остеобластов и остеокластов. Наиболее часто встречаются такие формы остеохондродисплазий:
- Болезнь Лери или мелореостоз;
- пикнодизостоз.
Мелореостоз чаще всего поражает мужчин, может развиться в любом возрасте. Характеризуется болезнь избыточным образованием эндостальной или периостальной кости. Процесс может происходит в двух зонах одновременно. Зарождается болезнь Лери с поражения нижних конечностей. Процесс может переходить на все суставы, отдельные кости таза, позвоночный столб, ребра и даже череп. Все пораженные кости довольно слабо изменены и деформированы, кортикальный стой утолщен, а костномозговая полость сужена неравномерно.
Мелореостоз может протекать совершенно бессимптомно продолжительное время, однако, при значительном уменьшении габаритов костномозговых лакун развивается болевой синдром в пораженной конечности. Нога при этом может укорачиваться или увеличиваться, развивается анкилоз сустав, нарушается гемопоэз.
Пикнодизостоз проявляется в виде карликовости и остеоскрероза. В основе заболевания лежит неравномерное, чрезмерное и очаговое периостальное развитие компактной кости. Развивается явная деформация скелета в виде:
- сколиоза;
- кифоза;
- гипоплазии ключиц;
- укорочении пальцевых фаланг;
- уменьшении длины предплечий.
В молочных зубах ребенка быстро развивается кариес, склеры глаз приобретают характерных болезни голубой оттенок. На продолжительности жизни пикнодизостоз не сказывается.
Фибродисплазия относится к редчайшему заболеванию, которое возникает в результате мутации определенного гена. На сегодняшний день в мире зарегистрировано 600 человек, страдающих от него. Патология характеризуется окостенением мышц, связок, сухожилий. Сначала поражается позвоночник, далее страдают другие кости организма.
Характеристика заболевания
Оссифицирующая фибродисплазия (ФОП) по МКБ 10 имеет номер М61.1, возникает из-за генетических мутаций, в результате видоизменения гена ACVR1. Известны случаи, когда поражаются несколько членов семьи, которые находятся в кровном родстве, братья и сестры. Встречается передача заболевания от одного поколения к другому, от отцов к детям.

Не следует путать данное заболевание с фиброзной дисплазией, при котором происходит замещение здоровой ткани кости на включение костной трабекулы. Фиброзная дисплазия относится к разряду опухолевых состояний, но не является раковой опухолью. Часто происходит перерождение в доброкачественное новообразование, онкология встречается редко.
У пациентов любой ушиб, небольшая ранка не вылечивается, на месте повреждения происходит образование костной ткани.
У новорожденного почти полностью определяется наличие заболевания, от которого одинаково страдают и девочки, и мальчики. Оно обнаруживается загнутым внутрь большим пальцем ноги и отсутствием сустава. Патология проявляется в первые 10 лет жизни больного, характеризуется внезапностью. Нижние конечности страдают в последнюю очередь.
Насколько быстро будет развиваться болезнь установить невозможно. Так как это зависит от количества полученных травм и перенесенных оперативных вмешательств.
Бывают случаи, когда врачи путают оссифицирующую фибродисплазию с онкологией, которая приводит к формированию костных образований. После их удаления происходит резкое усугубление ситуации и ускоренное развитие патологии.
Как развивается
Оссифицирующая фибродисплазия развивается по стадиям:
- На первой стадии образуется небольшой воспалительный очаг,
- На второй стадии патология начинает прогрессировать, воспаление вовлекается в ткани, расположенные около воспаленного места,
- На третьей стадии образуются бессосудистые уплотнения,
- Далее формируется хрящеобразование,
- Завершающей стадией является окостенение.
Причины
Оссифицирующая фибродисплазия начинает свое формирование еще в период внутриутробного развития, в результате изменения гена, расположенного во второй хромосоме. Из-за чего происходит мутация формы белка, отвечающего за развитие костной ткани.
Гетеротопические фибродисплазийные очаги обычно формируются в местах повреждения мягкой ткани, ожогов, укусов насекомых, порезов, ушибов. В данной области обнаруживается много лейкоцитов, являющихся фактором иммунного, воспалительного процесса.
Имеются пациенты, которые являются носителями гена, и при определенных обстоятельствах заболевание начинает прогрессировать. На скорость его проникновения оказывает воздействие активность гена. К провоцирующим факторам относятся:
- Хирургические вмешательства,
- Вакцинирование,
- Травмирование кожного покрова,
- Вирусная инфекция
Симптомы
Оссифицирующая фибродисплазия редко начинает развиваться сразу же после рождения ребенка. Врачи обращают внимание на нарушение сформированности пальчиков на ногах и руках. Никаких других внешних проявлений не наблюдается.
Оссификация начинается внезапно, порой без наличия объективной причины. Возраст ребенка может быть от 2-3 месяцев до нескольких лет. Чаще прогрессирование происходит в первые пять лет жизни. Симптомами заболевания считаются:
- Ранняя симптоматика проявляется формированием небольшого уплотнения, размером до 3 мм,
- При прогрессировании уплотнение увеличивается до 10 см,
- При пальпации наблюдается болезненность, иногда покраснение,
- Походка у пациента приобретает неуверенность, скованность,
- На лице отсутствуют какие-либо эмоции,
- Происходит уменьшение объема движений, что имеет связь с появлением новых уплотнений, приводя к полной обездвиженности,
- При прощупывании появляются боли в ногах,
- Нарушается чувствительность,
- Происходят сбои функционирования внутренних органов.
Наиболее очевидной клинической картиной заболевания считают окостенение мягкой ткани. Но возможно проявление следующих дефектов, говорящих о наличии оссифицирующей фибродисплазии:
- Короткий по отношении к остальным большой палец ноги, характеризующийся изогнутостью внутрь,
- Дефект в шейном отделе позвоночника,
- Короткий большой палец рук,
- Отставание в развитии,
- Глухота,
- Облысение,
- Искривление пятого пальца ног,
- Кривошея,
- Сколиоз,
- Тугая подвижность костного сочленения.
Известны случаи, когда уплотнения самостоятельно рассасываются, но в последствии образуются снова. После чего через 2-3 недели происходит их окостенение, в результате которого стает невозможным дальнейшее их рассасывание. У взрослого пациента прогрессирующая болезнь Мюнхеймера приводит к слиянию десятков очагов. Что в последствии вызывает глубокую инвалидизацию и летальный исход.

Диагностика
Диагностирование оссифицирующей фибродисплазии возможно после окончания форсированного костеобразования, которое происходит с момента рождения до 14 лет. Часто заболевание обнаруживается спустя 2 года после его развития. Диагностирование проводится следующими методами:
- Внешним осмотром,
- Рентгенографией,
- Генетической консультацией.
Во время внешнего осмотра у детей до 10-летнего возраста обнаруживаются многочисленные уплотнения, располагающиеся в разных частях тела. В более старшем возрасте определяется пониженная суставная подвижность, искривленный позвоночник, гетеротропические оссификационные очаги. Уплотнения можно увидеть даже невооруженным глазом.
В исключительных случаях может потребоваться гистологическое исследование, но оно проводится редко, чтобы исключить появления новых уплотнений.
Рентгенография полезна на начальном этапе оссифицирующе патологии, поскольку позволяет выявить дисплазию кости, клинодактилию. На поздних стадиях костные уплотнения появляются в мышечной ткани, сухожилиях. Если болезнь зашла далеко, то на рентгеновском снимке не возможно визуализировать внутренние органы.

Лечение
На сегодняшний день не существует методов, вылечивающих прогрессирующую оссифицирующую фибродисплазию. Но американскими учеными активно проводятся исследования гена, который влияет на развитие патологии. Разрабатываются генные блокаторы, которые способны остановить процесс мутации. Восстанавливается нормальное синтезирование белков за счет нейтрализации мутировавшего гена.
Для лечения оссифицирующей фибродисплазии используют следующие лекарственные препараты:
- Этидронат, который замедляет формирование костной ткани. Но ударная доза средства не только оказывает воздействие на патологические кости, но и приводит к разрушению основного скелета,
- Лекарственные средства, основанные на Ламидроновой кислоте, схожи своим действием с предыдущими препаратами, но не так активно влияют на основные кости,
- Преднизолон оказывает мощное противовоспалительное действие, снижает боль, устраняет опухоли. Курс лечения данным средством не должен превышать 2 недель. Иначе происходит ускорение роста костной ткани, подавление иммунитета, ухудшение зрения,
- Целебрекс замедляет воспаление,
- Метаксалон используется для снижения вспышек заболевания, приводит к ослаблению спазмов.
Улучшает состояние больных, страдающих от фибродисплазии, иммунотерапия Интерфероном. Ее действие направлено на замедление прогресса патологии, уменьшение числа уплотнений. Для облегчения самочувствия пациентам назначают обезболивающие и седативные препараты.
Наибольшую безопасность для больных представляет оперативное вмешательство по поводу устранения уплотнений и костного разрастания. После процедуры обнаруживается усиление процесса окостенения.
На начальном этапе оссифицирующей патологии больному назначается электрофорез на основе йодида калия. Противопоказаны пациентам УВЧ, массажи, парафинотерапия.
Людям с оссифицирующей фибродисплазией необходимо исключение хирургического вмешательства, любых видов инъекций для минимизации риска возникновения новых травм.
Данное заболевание имеет неблагоприятный исход. На продолжительность жизни больных оказывают влияние создание условий, исключающих травм, увечий, инфекционных заболеваний.

Фатальная семейная бессонница
Фатальная семейная бессонница — редчайшая наследственная болезнь, при которой человек погибает от неспособности заснуть. До сих пор она отмечалась лишь в сорока семьях по всему миру. Фатальная бессонница обычно проявляется между 30 и 60 годами (чаще всего — после 50 лет) и продолжается от 7 до 36 месяцев. По мере того как заболевание прогрессирует, пациент страдает от все более тяжелых нарушений сна, причем никакие снотворные ему не помогают. На первой стадии бессонница сопровождается паническими атаками и фобиями, на второй к ним прибавляются галлюцинации и повышенное потоотделение. На третьей стадии болезни человек полностью теряет способность спать и начинает выглядеть намного старше своих лет. Затем развивается деменция, и пациент погибает — как правило, от истощения или пневмонии.
Для болезни характерен аутосомно-доминантный тип наследования: то есть у нее нет носителей. Детям она передается от родителей с вероятностью 50% и только при условии, если кто-то из них болен. Мужчины и женщины болеют фатальной семейной бессонницей с одинаковой частотой. На сегодняшний день это заболевание считается неизлечимым.

Нарколепсия-катаплексия
Примерно в 80% случаев нарколепсии сопутствует катаплексия: внезапная эпизодическая потеря мышечного тонуса, которая повторяется регулярно. В легких случаях у пациента слегка отвисает нижняя челюсть и возникает ощущении слабости в коленях, но если состояние тяжелое, человек может внезапно упасть на ровном месте. Его сознание при этом остается ясным. Катаплексия развивается на фоне выраженных эмоциональных реакций: смеха, злости, страха или удивления, что делает подобное состояние особенно неудобным.
Причина возникновения болезни пока ясна не до конца, но в ряде случаев отмечалась мутация мутация нейромедиатора орексина (ген HCRT, 17q21), который регулирует процесс передачи возбуждающих сигналов в мозгу и влияет на сон и аппетит. Сигнальная система между орексинергическими и другими нейронами дает сбой, угнетается активность моноаминергических нейронов и снижается приток возбуждающих сигналов в кору.
Устранить нарколепсию-катаплексию невозможно, однако симптоматическое лечение есть. Пациенты начинают чувствовать себя лучше благодаря регулярному сну в строго определенное время и препаратам, активизирующим работу центральных адренергических систем.

Фибродисплазия
Фибродисплазия оссифицирующая прогрессирующая (ФОП) — редкое генетическое заболевание, при котором организм начинает формировать новые кости — оссификаты — в неположенных местах: внутри мышц, связок, сухожилий и других соединительных тканей. К их образованию может привести любая травма: ушиб, порез, перелом, внутримышечная инъекция или операция. Из-за этого удалять оссификаты нельзя: после хирургического вмешательства кость может только сильнее разрастись. Физиологически оссификаты не отличаются от обыкновенных костей и могут выдерживать значительные нагрузки, вот только находятся не там, где надо.
ФОП возникает из-за мутации в гене ACVR1/ALK2, кодирующем рецептор костного морфогенетического белка. Она передается человеку по наследству от одного из родителей, если он тоже болен. Быть носителем этого заболевания нельзя: пациент либо болен, либо нет. Пока ФОП относится к числу неизлечимых болезней, однако сейчас проводится вторая серия испытаний препарата под названием паловаротен, который позволяет заблокировать ген, ответственный за патологию.

Прогерия
Детская прогерия, или синдром Хатчинсона-Гилфорда, — заболевание, которым страдает один человек из миллионов. Организм детей с таким диагнозом стареет чрезвычайно быстро и рано: уже в подростковом возрасте пациенты выглядят и чувствуют себя, как старики. У них развивается множество старческих патологий, возникают нарушения в работе внутренних органов и систем, а кости, кожа, мышцы и сухожилия становятся слабыми и вялыми. При этом по уровню развития дети с прогерией не уступают сверстникам, а иногда и опережают их. Средняя продолжительность жизни людей, страдающих синдромом Хатчинсона-Гилфорда, — 13 лет. Как правило, причиной смерти становится инфаркт миокарда. Описан всего один случай, когда пациент с таким диагнозом дожил до 45 лет.
На данный момент это заболевание также считается неизлечимым, однако продлить жизнь пациентов и улучшить ее качество помогает разнообразное симптоматическое лечение и физическая активность, особенно плавание. Эти средства позволяют улучшить состояние кровеносной системы и суставов. Кроме того, используется гормон роста.

Синдром РОХХАД
Синдромом РОХХАД (Rapid-onset Obesity with Hypothalamic dysfunction, Hypoventilation and Autonomic Dysregulation) — чрезвычайно редкое заболевание, при котором человек начинает стремительно набирать вес и страдает от булимии, респираторных болезней, остановок дыхания во сне, альвеолярной гиповентиляции и кардиореспираторных остановок. Также для пациентов с таким диагнозом характерно отсутствие реакции на повышение в крови углекислого газа.
На сегодняшний день в мире зарегистрировано порядка 100 случаев возникновения этого расстройства. Обычно оно проявляется в возрасте до 10 лет (чаще всего около 3 лет) и, по всей видимости, имеет наследственную природу. Несмотря на проведенные на Западе исследования, этиология синдрома РОХХАД до сих пор не ясна. Считается, что он появляется из-за дисфункции гипофиза, которую вызывает генетическая мутация. Однако ученым еще только предстоит определить, какой именно процесс нарушается в этом случае.
Иконки: 1) Jamie Dickinson, 2) Jaclyne Ooi, 3) Abigail Cramer, 4) Luis Martins, 5) James Stone — from the Noun Project.
Читайте также:
