
Апоптоз и ревматоидный артрит
Данная диссертационная работа должна поступить в библиотеки в ближайшее время
Уведомить о поступлении
480 руб. | 150 грн. | 7,5 долл. ', MOUSEOFF, FGCOLOR, '#FFFFCC',BGCOLOR, '#393939');" onMouseOut="return nd();"> Диссертация, - 480 руб., доставка 1-3 часа, с 10-19 (Московское время), кроме воскресенья
Автореферат - бесплатно , доставка 10 минут , круглосуточно, без выходных и праздников
Рябков Вадим Александрович. Апоптоз лимфоцитов при ревматоидном артрите : автореферат дис. . кандидата медицинских наук : 14.00.39 / Ярослав. гос. мед. акад..- Ярославль, 2007.- 22 с.: ил. РГБ ОД, 9 07-3/775
Введение к работе
Актуальность проблемы
Ревматоидный артрит (РА) является одним из самых распространенных хронических воспалительных заболеваний суставов, которое поражает людей наиболее трудоспособного возраста, приводя к ранней инвалидизации и снижению продолжительности жизни больных (Е.Л. Насонов, 2002, И.А. Андрианова, В.Н. Амирджанова и соавт., 2006; О.М. Фоломеева, Ш.Ф. Эрдес, 2006).
Этиология РА достоверно неизвестна, а используемая патогенетическая терапия не оказывает достаточного эффекта у части больных, что определяет необходимость поиска новых подходов к лечению, основанных на данных о патофизиологии процесса (Е.Л. Насонов, 2004, C.M. Weyand, 2000).
В настоящее время большое внимание в патогенезе РА уделяется изучению апоптоза – генетически запрограммированной формы гибели клетки, обеспечивающей в организме клеточный гомеостаз (А.А. Ярилин, 1998; А.Н. Богданов, Т.А. Камилова и соавт., 2005; M. Schirmer, A.N. Vallejo et al., 1998; R.M. Pope, 2002).
Внимание к апоптозу как возможному механизму развития и поддержания патологического процесса при РА обусловлено современными представлениями о патофизиологии и патоморфологии заболевания. Поражение суставов в виде синовиальной гиперплазии позволяет предполагать нарушения программированной клеточной гибели синовиоцитов, приводящие к формированию паннуса, а местная инфильтрация различными клетками воспаления не исключает дефекты апоптоза лимфоцитов, поддерживающих хроническое течение болезни (А.Н. Богданов, Т.А. Камилова и соавт., 2006; K. Eguchi, 2001; H. Perlman, L.J. Pagliari et al., 2001; R.M. Pope, 2002; K. Itoh, H. Hase et al., 2004).
Менее изученным вопросом является процесс программированной клеточной гибели циркулирующих лимфоцитов, нарушение которого может вести к потере иммунологической толерантности в результате увеличения продолжительности жизни и накопления функционально активных аутореактивных Т-клеток (А.Н. Богданов, Т.А. Камилова и соавт., 2005; F.M. Bryl, A.D. Foey et al., 2002).
Продолжается изучение роли в нарушениях процесса апоптоза главных его участников – аспартат-специфических протеаз (каспаз (К)), среди которых выделяют активаторы провоспалительных цитокинов, активаторы эффекторных К и эффекторные К – непосредственные исполнители апоптоза (Е.Б. Владимирская, 2002; М.Ю. Григорьев, Е.Н. Имянитов, К.П. Хансон, 2003; А.А. Фильченков, 2003; J.M. Adams, 2003).
Проводятся исследования особенностей функционирования при РА основных регуляторов программированной клеточной гибели, в том числе наиболее изученного индуктора апоптоза Fas-лиганда (FasL), экспрессируемого на активированных Т-лимфоцитах, и его растворимой формы (sFasL) (A.N. Vallejo, M. Schirmer et al., 2000; O. Bohana-Kashtan, C.I. Civin, 2004). Фактор некроза опухоли-альфа (ФНО-), являющийся одним из основных участников патогенеза РА, в настоящее время рассматривается как регулятор апоптоза с разнонаправленными эффектами от апоптотической гибели до стимуляции клеточной пролиферации и дифференцировки (А.А. Ярилин, 1998). Продемонстрировано влияние интерлейкинов (ИЛ) как провоспалительных цитокинов на активность программированной клеточной гибели лимфоцитов синовиальной ткани и периферической крови при РА (K. Eguchi, 2001; H. Perlman, L.J. Pagliari et al., 2001).
На основании имеющихся данных последнее время предпринимаются попытки создания препаратов, влияющих на апоптоз, – блокаторов рецепторов, регулирующих программированную клеточную гибель, блокаторов ингибиторов апоптоза и средств, модифицирующих активность К (M.D. Smith, J.G. Walker, 2004; U. Fisher, K. Schulze-Osthoff, 2005).
В то же время активация апоптоза считается одним из механизмов патогенетического действия рекомендованных для лечения РА базисных противовоспалительных препаратов (БПВП) (Е.Л. Насонов, 2005; M. Cutolo, A. Sulli et al., 2001). Не исключается возможность прогнозирования эффективности базисных средств на основании результатов изучения программированной клеточной гибели (S. Herman, N. Zurgil et al., 2003). Более активное исследование влияния БПВП на апоптоз поможет объяснить различную чувствительность пациентов к имеющимся лекарственным средствам.
Несмотря на признанное участие периферических лимфоцитов в патогенезе РА опубликованные сведения, посвященные нарушениям апоптоза как причине накопления клона аутореактивных Т-клеток, весьма малочисленны. Имеющиеся данные отражают главным образом иммунологические характеристики программированной клеточной гибели in vitro без учета активности, течения заболевания и проводимой терапии.
С учетом вышеизложенного в работе необходимо продолжить изучение интенсивности апоптоза периферических лимфоцитов при различной активности РА и влияния проводимого лечения на программированную клеточную гибель.
Цель исследования
Изучить интенсивность апоптоза периферических лимфоцитов и оценить возможность использования показателей активности программированной клеточной гибели для прогнозирования эффективности проводимой терапии при ревматоидном артрите.
Задачи работы
Определить интенсивность апоптоза периферических лимфоцитов по активности каспаз-4, -6, -8 и количеству разрывов ДНК у больных ревматоидным артритом.
Сравнить результаты исследования активности апоптоза периферических лимфоцитов у больных ревматоидным артритом с показателями здоровых доноров и пациентов с остеоартрозом.
Оценить активность апоптоза периферических лимфоцитов у больных с впервые выявленным и ранее установленным ревматоидным артритом.
Оценить интенсивность программированной клеточной гибели периферических лимфоцитов в зависимости от активности ревматоидного артрита.
Сопоставить показатели активности апоптоза с основными клинико-лабораторными проявлениями ревматоидного артрита.
Определить влияние ФНО-, интерлейкина-2, интерлейкина-6, растворимого Fas-лиганда сыворотки крови на интенсивность апоптоза периферических лимфоцитов.
Изучить динамику апоптоза периферических лимфоцитов на фоне проводимой базисной терапии.
Оценить возможность использования показателей активности программированной клеточной гибели для прогнозирования эффективности терапии базисными противовоспалительными препаратами.
Научная новизна работы
В ходе исследования изучены особенности программированной клеточной гибели периферических лимфоцитов при ревматоидном артрите и остеоартрозе. Выявлены существенные различия в интенсивности апоптоза при этих заболеваниях.
Впервые методом флуоресценции определена активность каспаз и степень реализации программированной клеточной гибели в виде разрывов ДНК при ревматоидном артрите со сравнительной оценкой показателей при различных стадиях, длительности и активности заболевания. Обнаружена закономерная связь нарушений апоптоза периферических лимфоцитов с активностью ревматоидного артрита.
В работе впервые исследована динамика интенсивности программированной клеточной гибели периферических лимфоцитов на фоне проводимого лечения базисными противовоспалительными препаратами. Выявлено коррегирующее влияние эффективной базисной терапии в отношении ассоциированного с воспалительной активностью снижения реализации апоптоза.
Впервые оценена возможность использования показателей активности программированной клеточной гибели периферических лимфоцитов в качестве прогностических факторов эффективности лечения базисными противовоспалительными препаратами.
Практическая ценность работы
Полученные результаты позволяют оценить характер изменений активности апоптоза при ревматоидном артрите. В работе показана способность базисных противовоспалительных препаратов индуцировать программированную клеточную гибель периферических лимфоцитов. В ходе исследования выявлено, что количество разрывов ДНК и активность каспазы-4, определяемые в динамике методом флуоресценции как показатели интенсивности апоптоза, могут быть использованы в комплексе с традиционными клинико-лабораторными данными для оценки эффективности лечения базисными средствами при ревматоидном артрите.
Положения, выносимые на защиту
Для больных ревматоидным артритом характерна более высокая активность апоптоза периферических лимфоцитов, которая существенно отличается от таковой у здоровых.
Интенсивность программированной клеточной гибели периферических лимфоцитов при ревматоидном артрите зависит преимущественно от активности заболевания.
Эффективная терапия базисными противовоспалительными препаратами достоверно снижает активность ревматоидного артрита и усиливает апоптоз периферических лимфоцитов.
Апробация работы
Реализация результатов работы
Объем и структура диссертации
Ревматоидный артрит представляет собой системное иммуновоспалительное заболевание соединительной ткани, проявляющееся хроническим эрозивно-деструктивным полиартритом с преимущественно симметричным поражением суставов кистей и стоп и сопровождающееся у бо
Ревматоидный артрит представляет собой системное иммуновоспалительное заболевание соединительной ткани, проявляющееся хроническим эрозивно-деструктивным полиартритом с преимущественно симметричным поражением суставов кистей и стоп и сопровождающееся у большинства больных образованием особого вида аутоантител (ревматоидный фактор). Женщины болеют в 3–4 раза чаще мужчин, причем заболевание у них начинается обычно в возрасте 35–45 лет.
Болезненные, тугоподвижные суставы не только мешают выполнять самые простые повседневные движения, но и значительно нарушают ритм нормальной жизни.
Ревматоидный полиартрит — тяжелое системное заболевание, которое проявляется прогрессирующим воспалительным синовитом, симметрично поражающим периферические суставы конечностей.
В соответствии с рекомендациями Американской ревматологической ассоциации (1987) при диагностике ревматоидного артрита целесообразно придерживаться следующих критериев:
- утренняя скованность суставов или вблизи суставов длительностью не менее 1 ч до ее полного исчезновения;
- опухание мягких тканей (артрит) трех суставов или более, наблюдаемое врачом (т. е. в период осмотра или в анамнезе под наблюдением врача);
- опухание (артрит) проксимальных межфаланговых, пястно-фаланговых или запястных суставов;
- симметричное опухание (артрит) как минимум в одной паре суставов;
- ревматоидные узелки;
- наличие ревматоидного фактора;
- обнаружение при рентгенологическом исследовании эрозий и (или) периартикулярного остеопороза в суставах кистей и стоп.
При длительно текущем заболевании деформируются пораженные суставы. Помимо суставов, при ревматоидном полиартрите нарушаются околосуставные структуры (связки, сухожилия и др.), развиваются ревматоидный васкулит (поражение мелких сосудов), остеопороз, происходит поражение внутренних органов.
В пользу иммунопатологического/аутоиммунного характера воспалительного процесса при ревматоидном артрите свидетельствует обнаружение у большинства больных в сыворотке крови и синовиальном выпоте из пораженных суставов аутоантител — ревматоидного фактора.
Большое значение в развитии ревматоидного артрита имеет генетическая предрасположенность. Это подтверждается выраженной семейной агрегацией заболевания, наличием у части больных ревматоидным артритом антигенов II класса главного комплекса гистосовместимости HLA DR4 и Dw4 (с носительством которых связывают тяжелое течение артрита и быстрое прогрессирование эрозивных изменений суставов). Рассматривается роль наследственных или приобретенных нарушений Т-супрессорной регуляции иммунных реакций, недостаточной функции моноцитарно-макрофагальной системы. Пусковую роль в развитии ревматоидного артрита могут играть: хронические очаги инфекции, гормональная перестройка организма, пищевая аллергия, предшествующие травмы суставов, длительное воздействие влажного холода и физическое перенапряжение.
При хроническом воспалении у края суставного хряща в месте прикрепления к эпифизам суставной капсулы происходит разрушение и замещение паннусом участков субхондральной костной ткани с образованием эрозий. Иногда разрастающиеся грануляции проникают через субхондральную замыкательную пластинку в костную ткань. Продукты костно-хрящевой деструкции, в свою очередь, оказывают раздражающее воздействие на синовиальную оболочку, что способствует поддержанию в ней воспалительного процесса. Одновременно возникают воспалительные изменения в капсуле сустава, связках, синовиальной выстилке сухожилий и синовиальных сумках с последующим их склерозированием, приводящему к стойкому ограничению подвижности пораженных суставов, подвывихам и контрактурам. Иногда наблюдается некроз и разрыв сухожилий, синовиальных сумок. Нередко отмечается развитие вторичного остеоартроза.
Воспалительный процесс при ревматоидном артрите характеризуется неуклонным прогрессированием, темпы которого зависят от активности воспалительного процесса. Даже в периоды клинической ремиссии в синовиальной оболочке сохраняются признаки воспаления. Постепенно паннус разрушает хрящ на значительной поверхности эпифиза, а замещающая его грануляционная ткань соединяет между собой противоположные суставные поверхности и в последующем трансформируется вначале в фиброзную, а затем в костную ткань, что приводит к образованию соответственно фиброзного и костного анкилоза, обусловливающего полную неподвижность пораженных суставов.
При рентгенологическом исследовании суставов отмечается асимметричность эрозивных изменений с частым и ранним анкилозированием суставов запястий.
Течение серонегативного ревматоидного артрита менее тяжелое, в прогностическом плане более благоприятное, чем при серопозитивной форме заболевания: слабее выражены деструктивные (эрозивные) изменения и функциональные нарушения суставов, реже наблюдаются ульнарная девиация пальцев кистей, контрактуры и анкилоз (за исключением анкилоза суставов запястья). Вместе с тем серонегативный ревматоидный артрит хуже, чем серопозитивный, поддается терапии базисными и иммуносупрессивными препаратами. Чаще развивается вторичный амилоидоз.
При лечении ревматоидного артрита используются НПВП, в тяжелых случаях применяются глюкокортикостероиды, иммуносупрессоры, с целью профилактики остеопороза обычно назначают комбинацию препаратов кальция и витамина D.
Несмотря на все успехи фармакологии, ревматоидный артрит до настоящего времени остается тяжелым заболеванием с неблагоприятным прогнозом.
Приведем пример из личного опыта ведения больной с ревматоидным артритом, представляющим, по мнению автора, научный интерес.
Больная В., возраст — 71 год, обратилась к неврологу в августе 2006 г. с жалобами на боли в суставах кистей, коленных суставах, в пояснице. Боль в суставах беспокоила днем и ночью, обычно была выраженной, нередко мигрирующей, обусловливала ограничение всех активных и пассивных движений в пораженных суставах. Артралгия зачастую сочеталась с выраженной миалгией, оссалгией, болями в сухожилиях. Из анамнеза: проявления полиостеоартроза в течение 15 лет, дебют суставного синдрома воспалительного характера в июне 2002 г. (отек и воспаление в области суставов, фебрильная лихорадка, подтвержденная лабораторно активность воспалительного процесса). Амбулаторно (сначала периодически, в последние 3 года постоянно) получала НПВП (ибупрофен, индометацин, диклофенак, ацеклофенак). С 2004 г. регулярно принимала кальций D3 (1 г кальция в сутки).
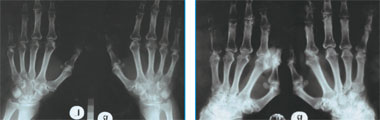 |
| Рис. 1. Рентгенограмма кистей больной В. 2003 г.: проявления полиостеоартроза, явных эрозий нет. Рис. 2. Рентгенограмма кистей больной В. 2006 г.: множественные кисты и эрозии, отрицательная динамика с 2003 г. |
Стационарно проходила лечение в городском ревматологическом центре г. Санкт-Петербурга (апрель 2003 г., июнь–июль 2006 г.) с диагнозом: ревматоидный артрит, серопозитивный, с системными проявлениями (субфебрилитет, амиотрофия, миалгии), активность 2-й степени, функциональная недостаточность суставов 1–2-й степени. Сопутствующая патология: ишемическая болезнь сердца, стенокардия II ФК. Гипертоническая болезнь 3-й степени, ОНМК (декабрь 2002 г.). Хронический бронхит вне обострения. Диффузный зоб второй стадии. Эутиреоз. Полиостеоартроз. Желчно-каменная болезнь.
С 2003 г. постоянно получает метотрексат в дозировке 2,5 мг 2 раза в неделю. Несмотря на проводимое лечение, суставной болевой синдром нарастал, учащались обострения, ухудшалось состояние костей (остеопороз, эрозии). Показательна рентгенография кистей (рис. 1–2).
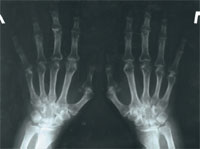 |
| Рис. 3. Рентгенограмма кистей больной В. 2007 г.: проявления полиостеоартроза, положительная динамика с 2006 г. |
С августа 2006 г. на фоне лечения метотрексатом неврологом был дополнительно назначен Структум в дозировке 500 мг 2 раза в день. Больная ориентирована врачом на длительный, до полугода, прием препарата. Некоторое уменьшение интенсивности (непрекращающегося в течение 2 последних месяцев) болевого синдрома наступило через 3 нед. Эпизоды обострений ревматоидного артрита были в течение 4 мес. К концу 5-го месяца комбинированной терапии (метотрексат + Структум) болевой синдром значительно снизился, уменьшилась утренняя скованность, спала отечность лучезапястных и коленных суставов.
В феврале 2007 г. была сделана рентгенография кистей (рис. 3). Результат сравнения с предыдущими рентгенографиями оказался более чем положительным (уменьшились эрозии костей). Прием структума продлен на 3 мес. По состоянию на май 2007 г. обострений суставного синдрома не наблюдалось в течение 4 мес, сохранялась небольшая отечность на лучезапястных (больше слева) и коленных суставах. С целью коррекции противоревматической терапии (снижение дозы метотрексата) больной рекомендована консультация в городском ревмоцентре.
И. Н. Бабурин
СПбНИПНИ им. В. М. Бехтерева, Санкт-Петербург
Полный текст:
- Аннотация
- Об авторах
- Литература
- Cited By
В статье суммированы данные литературы и собственных исследований авторов о молекулярно-клеточных механизмах апоптоза и их состоянии при ревматоидном артрите (РА). Нарушения процессов апоптоза при РА являются одной из причин гиперактивации синовиальных клеток, ведущей к усилению воспалительного процесса, гиперплазии синовиальной оболочки и прогрессированию заболевания в целом. Важнейшая особенность клеточного континуума пораженной синовии — сосуществование сразу двух механизмов контроля апоптоза: традиционного активационного, приводящего к прогрессированию воспаления и деструкции сустава, и ингибиторного, реализуемого через экспрессию проапоптотических молекул. Факторы апоптоза являются полезным инструментом для оценки прогноза РА и перспективной мишенью для фармакотерапии.
Дубиков Александр Иванович.
690080, Владивосток, бухта Патрокл, ул. Басаргина, 42В.
690002, Владивосток, просп. Острякова, 2.
690002, Владивосток, просп. Острякова, 2.
1. Матвеева НЮ, Матвеев ЮА, Калиниченко СГ, и др. Значение апоптоза энтероцитов при воспалительных заболеваниях кишечника. Бюллетень сибирской медицины. 2018;17(1):121-30.
2. Каминский ЮВ, Матвеев ЮА, Матвеева НЮ. Склеродермическая энцефалопатия. Тихоокеанский медицинский журнал. 2016;(2):108-12.
3. Malemud CJ, Pearlman E. Targeting JAK/STAT signaling pathway in inflammatory diseases. Curr Signal Transduct Ther. 2009;(4):201-21.
4. Jiao Y, Ding H, Huang S, et al. Bcl-XL and Mcl-1 upregulation by calreticulin promotes apoptosis resistance of fibroblast-like synoviocytes via activation of PI3K/Akt and STAT3 pathways in rheumatoid arthritis. Clin Exp Rheumatol. 2018 Sep-Oct;36(5):841-849. Epub 2018 Apr 13.
5. Dubikov AI, Kalinichenko SG. Small molecules regulating apoptosis in the synovium in rheumatoid arthritis. Scand JRheumatol. 2010; 39(5):368-72. doi: 10.3109/03009741003742771.
6. Korb A, PavenstKdt H, Pap T. Cell death in rheumatoid arthritis. Apoptosis. 2009 Apr; 14(4):447-54. doi: 10.1007/s10495-009-0317-y.
7. Wei XJ, Li XW, Lu JL, et al. MiR-20a regulates fibroblast-like synoviocyte proliferation and apoptosis in rheumatoid arthritis. Eur Rev Med Pharmacol Sci. 2017 Oct;21(17): 3886-3893.
8. Дубиков АИ, Белоголовых ЛА, Мед¬ведь ЕЭ. Апоптоз как механизм развития аутоиммунного воспаления в коленном суставе человека. Бюллетень экспериментальной биологии и медицины. 2004;(12): 641-4.
9. Wylie MA, Malemud CJ. Perspective: Deregulation of apoptosis in arthritis by altered signal transduction. Int J Clin Rheumatol. 2013;(8):483-90.
10. Vomero M, Manganelli V, Barbati C, et al. Reduction of autophagy and increase in apoptosis correlates with a favorable clinical outcome in patients with rheumatoid arthritis treated with anti-TNF drugs. Arthritis Res Ther. 2019 Jan 29;21(1):39. doi: 10.1186/s13075-019-1818-x.
11. Калиниченко СГ, Матвеева НЮ. Морфологическая характеристика апоптоза и его значение в нейрогенезе. Морфология. 2007;(2):16-28.
12. Grilo AL, Mantalaris A. Apoptosis: A mammalian cell bioprocessing perspective. BiotechnolAdv. 2019 May-Jun;37(3):459-475. doi: 10.1016/j.biotechadv.2019.02.012. Epub 2019 Feb 20.
13. Taylor RC, Cullen SP, Martin SJ. Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008 Mar; 9(3):231-41.
14. Hutcheson J, Perlman H. Apoptotic regu-lators and RA. CurrRheum Rev. 2008;(4):254-8.
15. Birbes H, El Bawab S, Obeid LM, Hannun YA. Mitochondria and ceramide: intertwined roles in regulation of apoptosis. Adv Enzyme Regul. 2002;42:113-29.
16. Fussenegger M, Bailey JE, Varner J. A mathematical model of caspase function in apoptosis. Nat Biotechnol. 2000 Jul;18(7): 768-74.
17. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2015 Apr 1;7(4). pii: a026716. doi: 10.1101/cshperspect.a026716.
18. Fava LL, Bock FJ. Caspase-2 at a Glance. J Cell Sci. 2012 Dec 15;125(Pt 24):5911-5. doi: 10.1242/jcs.115105.
19. Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017 Apr;16(4):273-284. doi: 10.1038/nrd.2016.253. Epub 2017 Feb 17.
20. Yamanishi Y, Boyle DL, Pinkoski MJ, et al. Regulation of joint destruction and inflammation by p53 in collagen-induced arthritis. Am J Pathol. 2002 Jan;160(1):123-30.
21. Taranto E, Xue JR, Lacey D, et al. Detection of the p53 regulator murine double-minute protein 2 in rheumatoid arthritis. JRheumatol. 2005 Mar;32(3):424-9.
22. Tang Y, Zhao W, Chen Y, et al. Acetylation is indispensable for p53 activation. Cell. 2008 May 16;133(4):612-26. doi: 10.1016/j.cell.2008.03.025.
23. You X, Boyle DL, Hammaker D, Firestein GS. PUMA-mediated apoptosis in fibroblast-like synoviocytes does not require p53. Arthritis Res Ther. 2006;8(6):R157.
24. Cha HS, Bae EK, Ahn JK, et al. Slug suppression induces apoptosis via Puma transactivation in rheumatoid arthritis fibroblast-like synoviocytes treated with hydrogen peroxide. Exp Mol Med. 2010 Jun 30;42(6): 428-36.
25. Firestein GS, Echeverri F, Yeo M, et al. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci U S A 1997 Sep 30;94(20): 10895-900.
26. Tong WW, Zhang C, Hong T, et al. Silibinin alleviates inflammation and induces apoptosis in human rheumatoid arthritis fibroblast-like synoviocytes and has a therapeutic effect on arthritis in rats. Sci Rep. 2018 Feb 19;8(1):3241. doi: 10.1038/s41598-018-21674-6.
27. Malemud CJ. The PI3K/Akt/PTEN/mTOR pathway: a fruitful target for inducing cell death in rheumatoid arthritis? Future Med Chem. 2015;7(9):1137-47. doi: 10.4155/fmc.15.55.
28. Boyle DL, Soma K, Hodge J, et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann Rheum Dis. 2015 Jun;74(6): 1311-6. doi: 10.1136/annrheumdis-2014-206028. Epub 2014 Nov 14.
29. Hutcheson J, Perlman H. BH3-only proteins in rheumatoid arthritis: potential targets for therapeutic intervention. Oncogene. 2008 Dec;27 Suppl 1:S168-75. doi: 10.1038/onc.2009.54.
30. Дубиков АИ. Ревматоидный артрит, апоптоз, оксид азота: новые аспекты патогенеза: монография. Владивосток: Издательство Дальневосточного университе-та; 2004. 132 с.
31. Дубиков АИ, Белоголовых ЛА, Мед-ведь ЕЭ. Роль апоптоза в патогенезе ревматоидного артрита и остеоартроза. Научно-практическая ревматология. 2005;43(1): 64-8. doi: 10.14412/1995-4484-2005-560
32. Дубиков АИ. Малые молекулы — регуляторы апоптоза клеток синовиальной оболочки у больных ревматоидным артритом. Научно-практическая ревматология. 2006;44(1):4-8.
33. Дубиков АИ. Апоптоз клеток синовиальной оболочки у больных ревматоидным артритом. Тихоокеанский медицинский журнал. 2008;(4):20-3.
34. Tu Y, Xue H, Xia Z, et al. Effect of different concentrations of dexamethasone on apoptosis and expression of Fas/FasL in human osteoarthritis chondrocytes. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi. 2012 May;26(5):536-41.
35. Tang Y, Luo J, Zhang W, et al. Tip60-dependent acetylation of p53 modulates the decision between cell cycle arrest and apoptosis. Mol Cell. 2006 Dec 28;24(6):827-39.
36. Krummel KA, Lee CJ, Toledo F, et al. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc Natl Acad Sci U S A. 2005 Jul 19;102(29):10188-93. Epub 2005 Jul 8.
37. Feng L, Lin T, Uranishi H, et al. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol Cell Biol. 2005 Jul;25(13):5389-95.
38. Дорошевская АЮ, Кондратовский ПМ, Дубиков АИ. Малые молекулы — ключевые участники патогенеза ревматоидного артрита. Клиническая медицина. 2012;(6): 12-8.
39. Cha HS, Rosengren S, Boyle DL, Firestein GS. PUMA regulation and proapoptotic effects in fibroblast-like synoviocytes. Arthritis Rheum. 2006 Feb;54(2): 587-92.
40. Al-Ghamdi A, Attar SM. Extra-articular manifestations of rheumatoid arthritis: a hospital-based study. Ann Saudi Med. 2009 May-Jun;29(3):189-93.
41. Blanchong CA, Olshefski R, Kahwash S. Large granular lymphocyte leukemia: case report of chronic neutropenia and rheumatoid arthritis-like symptoms in a child. Pediatr Dev Pathol. 2001 Jan-Feb;4(1):94-9.
42. Papadaki HA, Kritikos HD, Valatas V, et al. Anemia of chronic disease in rheumatoid arthritis is associated with increased apoptosis of bone marrow erythroid cells: improvement following anti-tumor necrosis factor-alpha antibody therapy. Blood. 2002 Jul 15;100(2): 474-82.
43. Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford). 2010 Sep;49(9): 1618-31. doi: 10.1093/rheumatology/keq045. Epub 2010 Mar 24.
44. Berliner N, Horwitz M, Loughran TP. Congenital and acquired neutropenia. Hematology Am Soc Hematol Educ Program. 2004:63-79.
45. Arend WP, Firestein GS. Pre-rheumatoid arthritis: predisposition and transition to clinical synovitis. Nat Rev Rheumatol. 2012 Oct; 8(10):573-86. doi: 10.1038/nrrheum.2012.134. Epub 2012 Aug 21.
46. Peng SL. Fas (CD95)-related apoptosis and rheumatoid arthritis. Rheumatology (Oxford). 2006 Jan;45(1):26-30. Epub 2005 Sep 13.
47. Zhang X, Feng H, Du J, et al. Aspirin promotes apoptosis and inhibits proliferation by blocking G0/G1 into S phase in rheumatoid arthritis fibroblast-like synoviocytes via downregulation of JAK/STAT3 and NF-к B signaling pathway. Int J Mol Med. 2018 Dec; 42(6):3135-3148. doi: 10.3892/ijmm.2018.3883. Epub 2018 Sep 17.
48. Амирджанова ВН, Кайгородцева ЕЮ, Савенкова НА, Насонов ЕЛ. Качество жизни как критерий эффективности терапии базисными противовоспалительными и генно-инженерными препаратами: результаты международных и российских исследований. Научно-практическая ревматология. 2009;47(2):54-66. doi: 0.14412/1995-4484-2009-460
49. Newton K. RIPK1 and RIPK3: Critical regulators of inflammation and cell death. Trends Cell Biol. 2015 Jun;25(6):347-53. doi: 10.1016/j.tcb.2015.01.001.Epub 2015 Feb 4.
50. Assmann G, Wagner AD, Monika M, et al. Single-nucleotide polymorphisms p53 G72C and Mdm2 T309G in patients with psoriasis, psoriatic arthritis, and SAPHO syndrome. Rheumatol Int. 2010 Aug;30(10): 1273-6. doi: 10.1007/s00296-009-1136-8. Epub 2009 Sep 25.
51. Malemud CJ. Defective T-Cell Apoptosis and T-Regulatory Cell Dysfunction in Rheumatoid Arthritis. Cells. 2018 Nov 22; 7(12). pii: E223. doi: 10.3390/cells7120223.
52. Cosme-Blanco W, Shen MF, Lazar AJ, et al. Telomere dysfunction suppresses spontaneous tumorigeesis in vivo by initiating p53-dependent cellular senescence. EMBO Rep. 2007 May;8(5):497-503. Epub 2007 Mar 30.
53. Van Nguyen T, Puebla-Osorio N, Pang H, et al. DNA damage-induced cellular senescence is sufficient to suppress tumorigenesis: a mouse model. J Exp Med. 2007 Jun 11; 204(6):1453-61. Epub 2007 May 29.
54. Hong LZ, Zhao XY, Zhang HL. P53-mediated neuronal cell death in ischemic brain injury. NeurosciBull. 2010;26(3):232-40.

Контент доступен под лицензией Creative Commons Attribution 4.0 License.
Читайте также:
