
Упражнения при спинальной амиотрофии
Начнем, пожалуй, с определения термина. Вообще, медикам достаточно легко понять друг друга, так как они знают латынь. А как быть всем остальным, нормальным людям? Остается, к сожалению, спрашивать врачей. Однако врачи не всегда хотят быть ходячими справочниками и это понятно — у них куча другой работы. Но объяснение все-таки можно найти — именно для этого и существует наш блог.
Итак, сначала оригинальное определение: спинальные амиотрофии — группа наследственных, чаще аутосомно-рецессивных, заболеваний мотонейронов спинного мозга, характеризующаяся мышечной слабостью, подергиваниями мышц. А теперь переведем на русский:
Что имеем в остатке? Спинальные амиотрофии — группа наследственных заболеваний двигательных нейронов спинного мозга, которые передаются с неполовыми хромосомами и характеризуются мышечной слабостью (нарушением питания), подергиваниями мышц. Не характерны нарушения чувствительности, координации и глазодвигательные нарушения (именно потому, что заболевание поражает мотонейроны в спинном мозгу).
Вы заметили, что я говорю о заболеваниях. Да, их существует несколько типов:
- спинальная амиотрофия Верднига-Гоффмана 1 — очень раннее начало (до 6 месяцев) и, к сожалению, быстрый летальный исход — к 2-5 годам;
- спинальная амиотрофия Верднига-Гоффмана 2 — начало в раннем детстве (где-то около года-полутора), течение средней тяжести, средняя продолжительность жизни с таким типом заболевания — 10-30 лет;
- спинальная амиотрофия Кульдберга-Веландер — начинается у подростков, благоприятный прогноз — срок жизни больных более 40 лет;
- спинальная амиотрофия Кеннеди — проявляется у взрослых в сочетании с увеличением молочных желез. Этот тип сцеплен с х-хромосомой (то есть достается в наследство девочкам — от матери или отца, мальчикам — от матери);
В мышцах — картина, типичная для нейрогенных (т.е. вызванных поражениями нервов и нейронов) амиотрофий — соседство здоровых мышечных волокон и пораженных. Симптомы же болезни следующие:
- основной симптом болезни — постепенно нарастающая слабость и атрофия мышц. Процесс начинается с туловища и проксимальных (ближних к туловищу, например, плечи и бедра) отделов
конечностей, распространяясь потом симметрично на остальные группы мышц. Особенно часто болезнь затрагивает длинные мышцы спины. - Снижен мышечный тонус – часто атрофии мышц маскируются хорошо (или избыточно) развитой подкожной жировой клетчаткой. Однако если сделать электромиографию (это то же, что и электрокардиография, только для скелетных мышц), то мы увидим, что природа поражения мышц связана именно с нервами (неврогенная).
- Часто наблюдаются подергивания мышц и отсутствуют сухожильные рефлексы (так как нарушен путь прохождения импульса в спинном мозге).
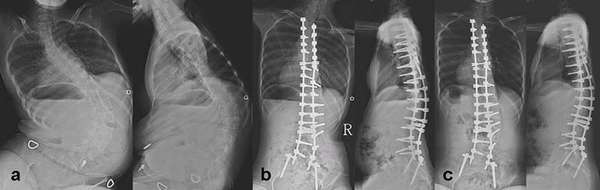
- Также характерные симптомы для спинальной амиотрофии — кифосколиоз (искривление позвоночника вбок и кзади), мышечные контрактуры (стойкое ограничение подвижности мышцы, она не может сокращаться и расслабляться) и нарушения дыхания из-за поражения межреберных мышц.
- Очень характерный симптом спинальной амиотрофии – истончение длинных костей (бедренная, плечевая и т.д.), выявляемое на рентгенограмме.
Спинальная амиотрофия почти всегда начинается на первом году жизни ребенка, чаще в первые 6 месяцев. К сожалению, течение быстро прогрессирует – 80 детей умирают до 4-летнего возраста, 56% — на первом году жизни. Есть и случаи, когда дети доживали до 20 лет, будучи тяжелыми инвалидами. Так как заболевание имеет генетическую природу, возраст начала болезни и ее длительность в каждой семье постоянны.
Спинальную амиотрофию необходимо отличать от следующих заболеваний:
- Острый полиомиелит – для него характерно острое начало с лихорадкой, параличи не симметричны и отсутствует прогрессирование процесса.
- Миопатии – тоже прогрессирующее наследственное заболевание мышц, но связано оно с нарушением обмена веществ в мышечной ткани. При миопатиях нет подергиваний мышц, не такое быстрое течение и другая электромиографическая картина.
- Врожденная миатония (нарушение тонуса мышц) – вот от нее спинальную миотрофию отличить не так просто. Основной признак врожденной миатонии – распространенная и сильно выраженная гипотония (снижение тонуса) мышц. Миаотния отличается от спинальной амиотрофии доброкачественным течением и отсутствием подергиваний мышц. Отличить эти два
заболевания друг от друга помогает биопсия (взятие образца ткани) мышц.
У нас, у медиков, есть два типа лечения – симптоматическое и патогенетическое. Первое направлено только на облегчение симптомов и не лечит саму болезнь (хотя косвенно – за счет улучшения физического и психического состояния – может и помогать в лечении болезни), например, чай с малиной при гриппе или простуде. Второе же направлено именно на лечение самой болезни, причем симптомы (боль и т.д.) могут исчезнуть и не сразу.
Так вот, к сожалению, патогенетического лечения спинальной амиотрофии пока не существует. Лечение только симптоматическое – хорошее питание, регулярная лечебная физкультура для спины и раннее проведение мероприятий, предотвращающих появление контрактур в мышцах.

Из всех существующих и известных науке спинальных мышечных атрофий амиотрофия Верднига-Гоффмана является наиболее тяжелой разновидностью.
Распространенность этого заболевания находится сейчас на отметке 1 случай на 7-11 тыс. новорожденных малышей.
Ген, который вызывает это заболевание, имеется у каждого 50-го человека.
Однако, благодаря аутосомно-рецессивному типу наследования, проявляется нарушение у ребёнка только тогда, когда эта генетическая информация имеется у обоих родителей.
Поэтому вероятность того, что малыш родится с патологией, в данном случае составляет около 25%.
Можно ли справиться с таким заболеванием или хотя бы остановить прогрессию симптомов расскажем в этой статье.
Что такое амиотрофия Верднига-Гоффмана?
Спинальная амиотрофия 1-го типа или, по-другому, спинальная амиотрофия Верднига-Гоффмана - это особое заболевание нервной системы, передающееся по наследству (чаще всего от обоих родителей). Эта патология характеризуется наличием мышечной слабости практически во всей мышечной системы организма. Ребёнок, страдающий от такого заболевания, не может самостоятельно сидеть, передвигаться и обслуживать себя.
К большому сожалению в мире не существует лекарства от этого типа заболевания. Максимум, что могут предложить врачи в наше время - дородовая диагностика. Такое обследование помогает избежать рождения больного малыша в семье.
Свое название патология получила от двух учёных, впервые описавших её в конце 19 в. В настоящее время под понятием спинальной амиотрофии понимается несколько форм болезни, отличающихся клинически. Но все они при этом связаны одним и тем же генетическим дефектом, которым обладают родители ребёнка.
Спинальная амиотрофия имеет несколько форм и разновидностей, каждая из которых отличается возрастом появления характерных симптомов, тяжестью протекания заболевания и продолжительностью жизни пациентов.
Обычно эта патология приводит к инвалидности, поскольку нарушается двигательная система организма, и пациент не способен ни самостоятельно передвигаться, ни самостоятельно себя обслуживать. При тяжелых клинических ситуациях может понадобиться постоянный врачебный контроль в повседневной жизни.

Передвигаться такому больному помогают инвалидные кресла, ходунки, костыли, трости. К смертельному исходу такое заболевание может привести только в том случае, когда появляются осложнения со стороны дыхательной и сердечно-сосудистой системы (при пневмониях и сердечной недостаточностью).
Под воздействие патологии не попадают чувствительные нервные волокна, поэтому у ребёнка сохраняются все виды чувствительности. Не страдают также интеллект и ментальные функции, так при обучении ребёнок совершенно нормально воспринимает и усваивает информацию.
В зависимости от возраста, при котором появились характерные симптому заболевания, амиотрофия Верднига-Гоффмана делится на несколько видов:
- Врожденная форма патологии. Примерный возраст появления изменений: от 0 до 6 месяцев. Обычно характеризуется слабым внутриутробным шевелением плода. При врождённой форме мышечная гипотония наблюдается с первых дней жизни малыша. В течение короткого времени происходит угасание глубоких рефлексов: ребёнок слабо кричит, плохо сосет молоко матери или соску, не может держать головку. Иногда случается так, что эти симптомы проявляются несколько позже, поэтому малыш может учиться держать головку и сидеть, но, поскольку имеется нарушение, эти навыки у него не разовьются. Также врожденная форма может сопровождаться бульбарными нарушениями, снижением глоточного рефлекса и фасцикулярными подергиваниями языка. Врожденная форма считается наиболее злокачественной и часто может сочетать в себе ещё и олигофрению, деформации грудной клетки, 3 и 4 степени сколиоза. Быстрая обездвиженность и парез дыхательной системы приводит к дыхательной недостаточности и впоследствии к летальному исходу;
- Ранняя детская форма. При данной разновидности патологии первые симптомы могут проявиться после 6 месяцев. К этому моменту дети имеют нормальное физическое и психическое развитие. Они начинают потихоньку приобретать первые естественные навыки, вроде умения держать головку, стоять, садиться и переворачиваться. В большинстве случаев, при наличии данного типа заболевания, дети так и не научатся ходить. На начальной стадии возникают парезы в нижних конечностях, затем довольно быстро они развиваются в верхних конечностях и во всей мускулатуре. Наступает мышечная гипотония, угасают глубокие рефлексы, может проявиться тремор пальцев, непроизвольные мышечные сокращения. На более поздних этапах ко всем симптомам добавляются бульбарные нарушения, дыхательная недостаточность (прогрессирующая). Эта форма заболевание протекает медленнее, чем врожденный тип. Больные могут дожить вплоть до 15 лет;
- Амиотрофия Кугельберга-Веландера. Самая доброкачественная из всех форм спинальной амиотрофии. Симптомы проявляются после 2-х лет, иногда в период между 15-ю и 30-ю годами. При данной форме не встречается психической задержки развития, довольно длительное время пациенты способны двигаться самостоятельно. Многие доживают до глубокой старости на полном самообслуживании.
Факторы риска и причины заболевания
Поскольку спинальная амиотрофия Верднига-Гоффмана является наследственным заболеванием, то и причины её возникновения кроются в генетическом коде обоих родителей пациента. Проблема скрывается в пятой хромосоме, которая подвергается генетической мутации.
Мутирует ген, который отвечает за производство SMN белка. В здоровом организме синтез этого белка обеспечивает нормальное развитие двигательных нейронов. Если он подвергся мутации, то двигательные нейроны начинают разрушаться, что приводит к нарушению передачи импульса от нервного волокна к мышце. Как следствие - мышца не функционирует. Именно поэтому возникает двигательная атрофия и невозможность нормально передвигаться.

Ген с нарушениями имеет аутосомно-рецессивный тип наследования. Это значит, что для того, чтобы болезнь развилась, требуется совпадение двух мутировавших генов от обоих родителей. Т.е. по сути и отец, и мать должны быть носителями гена с патологией.
При этом они не больны, ведь у них имеется доминантный здоровый ген (это также обусловлено парностью генов). Если и у отца, и у матери малыша имеется ген с патологией, то риск того, что ребёнок родится с нарушениями составляет 25 %.
Видео: "Что такое спинальная мышечная атрофия?"
Методы диагностики заболевания
Когда речь заходит о диагностике такого рода заболевания, то нужно учесть, что для невролога, который будет проводить обследование, очень важен возраст появления первых симптомов у малыша.
Также важна динамика развития симптомов, данные неврологического статуса (т.е. наличие/отсутствие двигательных нарушений периферического типа на фоне нормальной чувствительности), наличие/отсутствие дополнительных врожденных аномалий и костных деформаций (склиоза, кифоза, лордоза, кривошеи).
Врожденный тип заболевания может быть выявлен неонатологом. Обследование проводится с миопатиями, мышечной дистрофией (прогрессирующей), боковым амиотрофическим склерозом, полиомиелитом, ДЦП и др. Если диагноз требует максимально точного подтверждения, то применяется также электронейромиография (исследования нервно-мышечного аппарата).

Итоговый диагноз устанавливается только после получения данных о биологии мышц и исследования генетической ситуации. Изучение ДНК-анализа позволяет генетикам выяснить гетерозиготное носительство генной аберрации (важно при планировании следующей беременности). Проводится также количественный анализ числа генов локуса СМА (позволяет высчитать наличие патологического гена у родителей.
Анализ ДНК, сделанный в дородовый период, может помочь снизить вероятность рождения ребёнка с патологией Вердинга-Гоффмана. Но сложность здесь заключается в том, что для получения ДНК-материала применяются инвазивные методы пренатальной диагностики (биопсия хориона, кордоцентез, амниоцентез).
Если внутриутробно болезнь подтвердится, то это выступит показанием к искусственному прерыванию текущей беременности.
Лечение заболевания
Современная медицина, к большому сожалению, ещё не разработала препараты, способные справиться с генетическими мутациями разного рода, поэтому лечебного курса, способного победить амиотрофию Верднига-Гоффмана, не существует. Есть ряд мероприятий, которые могут замедлить прогрессирующую атрофию мышц, но чудес ждать не приходится.
Медикаменты способны только ослабить состояние больного, несколько поддержать и подпитать его нервную систему, но победить причину развития заболевания они не могу.
При спинальной амиотрофии применяют следующие препараты:
- Церебролизин, Цитофлавин, Глутаминовая кислота, АТФ, хлорид Карнитина, Метионин, Калия оротат и др. - эти препараты несколько улучшают метаболизм мышц и нервных тканей, поэтому их прописывают для периодического курсового приёма;
- Витамины В (Нейровитан, Мильгамма);
- Анаболические стероиды (Ретаболил, Неробол);
- Прозерин, Нейромидин, Дибазол - эти препараты способны несколько улучшить мышечную проводимость.

Амиатрофия Верднига-Гоффмана относится к орфанным (редким) патологиям, частота встречаемости которой 1:6000. Характеризируется крайне тяжелым течением и быстрым прогрессированием, приводящим к стойким и выраженным деформациям опорно-двигательного аппарата и летальному исходу.
- Что это?
- Причины заболевания
- I тип амиотрофии
- II тип
- III тип
- IV тип
- Диагностика
- Методы лечения
- Прогноз: сколько живут пациенты?
- Что нужно запомнить?
Что это?
Спинальная амиотрофия – генетическое заболевание, при котором обнаруживается мутации в 5 хромосоме в генах SMN1 и SMN2. Эти гены отвечают за сохранение и нормальное функционирование мотонейронов – нервов, которые отправляют импульс к скелетной мускулатуре. В результате отсутствия стимуляции мышц они атрофируются. У больных отмечается дегенеративные процессы в спинном мозге: разрушаются двигательные нейроны, нарушается функционирование передних рогов спинного мозга. В головном мозге изменения происходят в двигательных ядрах.
При мышечной амиотрофии Верднига мутирует ген SMN1, который препятствует гибели мотонейронов. Ген SMN2 выполняет эту функцию только частично и его возможности быстро истощаются. Течение и тяжесть патологии зависит от локализации и объема поражения в генном аппарате.

Фото 2
Спинальная амиотрофия вне зависимости от ее типа и формы – генетическое врожденное заболевание. Зачастую, первые признаки патологии обнаруживаются в младенческом возрасте либо еще во время беременности матери ребенка.
Одинаково распространена как у мужчин, так и женщин. Основное условие ее развития – наличие дефектного гена у обоих родителей, которые могут быть здоровыми и являются лишь носителями дефектной хромосомы.
В результате нарушения проводимости нервных импульсов от мотонейронов к мускулатуре происходит атрофия мышц нижних и верхних конечностей, диафрагмы, органов ЖКТ, сердца и другие. На фоне этого деформируются кости и суставы. Наиболее опасным проявлением является нарушения дыхательной функции и работы сердца.
В зависимости от тяжести течения, локализации дефектов и клинических проявлений выделяют 4 формы амиотрофии. Пациенты с любой формой амиотрофии являются инвалидами. Они не способны самостоятельно себя обслуживать, и нуждаются в постоянном уходе и медицинском наблюдении.
Причины заболевания
Основная причина развития спинальной амиотрофии – генетическая мутация у родителей и передача рецессивного гена плоду. Но, что привело к ее возникновению, ученым еще не известно. Мутация может произойти спонтанно, без видимых на то причин. Предполагаемыми причинами болезни являются следующие факторы:
- вирусные заболевания;
- нарушение экологии;
- прием во время беременности наркотиков, алкоголя, курение;
- воздействие терратогенных факторов;
- ионизирующее излучение.
В большинстве случаев родители узнают о том, что являются носителями опасного гена, только после рождения ребенка с диагнозом.
При некоторых вариантах амиотрофии толчком для прогрессирования мышечной дистрофии могут быть перенесенные вирусные заболевания, инсоляция, гормональный сбой.
I тип амиотрофии
Наиболее злокачественный и распространенный тип патологии – амиотрофия Верднига-Гоффмана. Диагностируется у детей раннего возраста либо внутриутробно. У детей после рождения отмечается снижение и угасание всех рефлексов. Им сложно сосать грудь, из-за чего часто возникает необходимость кормить ребенка через зонд.
Мышечная гипотония приводит к слабости шейных мышц – дети не могут научиться держать головку, самостоятельно переворачиваться, сидеть, стоять и ходить. У большинства детей возникают трудности с глотанием. В большинстве случаев амиотрофию диагностируют в течение 6 месяцев после рождения. Родители обращают внимание на вялость ребенка и малоподвижность, прогрессирующее снижение мышечного тонуса. Также младенцы плохо набирают вес.
Характеризируется поражением диафрагмы, которая участвует в дыхательном акте. Многие пациенты самостоятельно не дышат. Из-за выраженной дыхательной недостаточности многие дети не доживают до года. Продлить им жизнь удается с помощью ИВЛ и питания через зонд. 95% детей умирают до 4 лет.
Патология сопровождается прогрессирующими деформациями опорно-двигательной системы.
Помимо поражения мышц и костей, часто выявляется отставание в умственном развитии.
Справка. Заподозрить диагноз можно еще во время беременности матери, когда отмечается слабое шевеление плода, нарушение сердечной деятельности, отставание в развитии.
II тип
Промежуточная амиотрофия. Диагностируется у детей 3 месяцев—1,5 года. В более раннем возрасте диагноз трудно установить из-за особенностей мышечной системы у детей. Из-за выраженной мышечной гипотонии и снижения сухожильных рефлексов младенцы не могут научиться ползать и ходить. Лишь в 25% возможно, что ребенок сможет самостоятельно сесть.
Сопровождается поражением костно-суставной системы. У многих искривляется позвоночник, деформируется грудная клетка, атрофируются суставы рук и ног. Родители отмечают похудение ребенка либо прекращение набора веса.
Особенность этого типа амиотрофии – возможное внезапное прекращение ее развития. Несмотря на ремиссию, утратившиеся способности практически невозможно восстановить. Прогрессирование болезни может возобновиться либо ускориться после перенесенного ОРВИ или ОРЗ и других заболеваний, снижающих иммунитет. Из-за атрофии диафрагмы нарушается работа легких, ослабевает работа легких, из-за чего возникает одышка и тахикардия. Вовлечение в процесс мускулатуры органов пищеварения, бульбарные нарушения приводят к неспособности самостоятельно принимать пищу.
III тип
Болезнь Кугельберга-Веландера – вариант поздней амиотрофии. При данной форме диагноза первые признаки возникают после двух лет, либо во взрослом возрасте. Дети, которые уже умеют ходить, внезапно становятся неловкими, жалуются на боль в ногах при хождении. Они слишком часто падают, неуверенно ходят и практически перестают бегать. Многие родители замечают изменения походки и внезапно появившуюся неуклюжесть. Вначале развития патологии поражаются мышцы ног, затем атрофируются мышцы верхних конечностей и других частей тела. Из-за снижения объема мышц также отмечается снижение веса.
У некоторых больных может наступить период ремиссии, в который прекращается прогрессирование диагноза. Этот период может продлиться от нескольких недель, до нескольких десятков лет.
Нарушение дыхательной функции наиболее опасное проявление атрофии мускулатуры.
Со стороны психики и умственного развития нарушения не выявляются.
IV тип
Первые признаки и симптомы появляются в 30-50 лет. Основными жалобами является слабость мышц, тремор. Из-за атрофии появляются контрактуры в области суставов, с ограничением их подвижности. Больные резко худеют.
Часто сопровождается искривлением позвоночника и деформацией грудной клетки.
Первыми в процесс вовлекаются мышцы ног, после постепенно вовлекаются руки. Как правило, дыхательная и глотательная функция не нарушена.
Большинство пациентов самостоятельно ходят, но хромают, испытывают боль и дискомфорт. В редких случаях прогрессирование амиотрофии приводит к потере способности ходить, и больные вынуждены передвигаться на инвалидной коляске.
Диагностика
Основным методом диагностики является генетическое исследование. Для проведения тестирования используют кровь. В некоторых случаях для подтверждения диагноза проводят биопсию пораженных тканей.
Для определения тяжести болезни, объема поражений и нарушения функционирования мышечной системы проводят электромиографию.
Также обследовать необходимо и других близких родственников пациента.
Важно! Для пренатальной диагностики исследуют околоплодные воды. Такой метод применяется только при наличии симптомов и генетической предрасположенности у родителей.
Методы лечения
Генетическое заболевание является неизлечимым и практически не поддается коррекции. При прогрессирующем течении практически невозможно предотвратить и остановить мышечную атрофию. Несмотря на развитие генной инженерии, ученым не удалось создать лекарство, способное устранить генетический сбой.
Лечение направлено на поддерживание жизненно важных функций организма, облегчение состояния больного и предотвращение деформации скелета. Для этого применяют следующие методы:
- искусственная вентиляция легких – показана при дыхательной недостаточности;
- физиопроцедуры – массаж, ЛФК, электрофорез, лечебные ванны и другие;
- ношение корсета и других ортопедических приспособлений;
- витаминотерапия;
- применение ноотропов и препаратов, улучшающих питание нервной ткани: актовегин, трентал, церебролизин и другие.
Паллиативная терапия направлена на облегчение состояние тяжелобольных пациентов.
Ученые всего мира пытаются создать препарат для лечения патологии. Известно, что уже разработано несколько препаратов, которые возможно сумеют помочь пациентам. Они находятся на стадии клинических испытаний и их эффективность и безопасность для организма только изучается.
Родителям детей с тяжелым, неподдающимся лечению, диагнозом, для рождения здорового ребенка необходимо планировать беременность с репродуктологом. Искусственное оплодотворение, использование донорской спермы или яйцеклетки, экстракорпоральное оплодотворение – одни из способов родить здорового ребенка носителям дефектного гена.
Прогноз: сколько живут пациенты?
Спинальная амиотрофия крайне тяжелое заболевание, которое существенно ухудшает качество жизни больных и является одной из причин младенческой смерти. Пациенты с таким диагнозом являются инвалидами и зачастую не в силах самостоятельно себя обслуживать.
Многие пациенты не могут ходить и стоять, передвигаясь с помощью инвалидной коляски. При поражении рук больные не способны делать элементарных вещей: есть, держать небольшие предметы, читать, умываться и прочее. Они нуждаются в дополнительном уходе и постоянном медицинском наблюдении. При тяжелом течении они дышать лишь с помощью аппарата ИВЛ и питаются через зонд.
Продолжительность жизни во многом зависит от формы заболевания. Наиболее тяжелой является амиотрофия Верднига, при которой больные дети редко достигают 4 лет. Пациенты со вторым типом болезни редко доживают до совершеннолетия. Наиболее благополучным являются второй и третий тип болезни, при которых пациенты достигают зрелого возраста.
В настоящее время спинальная амиотрофия относится к неизлечимым заболеваниям, часто приводит к летальному исходу.
Спинальная мышечная атрофия является генетическим заболеванием, которое способно развиваться как в младенческом, так и во взрослом возрасте. Каждому человеку важно иметь представление о признаках патологии, это поможет вовремя обратиться к врачу и избежать неблагоприятных осложнений.
Что такое спинальная мышечная атрофия
Спинальная мышечная атрофия, или СМА (код по МКБ 10 – G 12), – это термин, который объединяет группу патологий, общей чертой которых является поражение двигательных нейронов спинного мозга. Данный процесс обуславливает клиническую картину заболеваний, которая характеризуется утратой двигательной активности и развитием в связи с этим различных осложнений.
Имеется несколько видов СМА, которые отличаются сроками начала появления признаков болезни и прогнозом относительно продолжительности жизни.
Причины развития
Причины возникновения заболеваний данной группы – генетические дефекты. Они приводят к разрушению структур нервных клеток, что проявляется атрофией мышечной ткани. Существует несколько типов СМА, которые наследуются как доминантным, так и рецессивным путем.
Классификация и симптомы по типовым различиям
Существует спинальная мышечная атрофия 1, 2 и 3 типа, также имеется 4-й вид заболевания. Выглядят они следующим образом.
Спинальная мышечная атрофия Верднига-Гоффмана относится к 1 типу патологии, является самым неблагоприятным видом заболевания, развивается в течение первого полугода жизни ребенка. У таких больных имеются проблемы с дыханием, проглатыванием пищи, задержки в моторном развитии. У детей не развиваются двигательные навыки, они не могут держать голову, сидеть, переворачиваться.
Пища часто попадает в дыхательные пути, что приводит к застойному воспалению легких, это нередко становится причиной гибели больных.
Важно отметить, что СМА 1 типа нередко сопровождается другими врожденными аномалиями. Пациенты редко доживают до возраста 3 лет.
Срок начала амиотрофии 2 типа – 6 месяцев – 2 года. До этого периода дети развиваются нормально, с началом заболевания возникают жалобы на мышечную слабость, которая постепенно прогрессирует. Мышцы понемногу истончаются, что нарушает процесс ходьбы. У пациентов нередко диагностируются нарушения осанки, патологии суставов, могут быть дыхательные инфекции. В целом прогноз достаточно благоприятный.
Атрофия мышц 3 типа, или болезнь Кукельберга-Веландера, чаще развивается в подростковом возрасте. Пациенты начинают чувствовать слабость в нижних конечностях, которая неуклонно прогрессирует и приводит к тому, что больные перестают передвигаться. Постепенно вовлекаются мышцы рук и лица, присоединяются деформации позвоночника и суставов.
Данный тип заболевания развивается только у взрослых людей, после 35 лет. Атрофия 4 вида довольно благоприятная, в патологический процесс вовлекаются нижние конечности.
По мере прогрессирования нарастает слабость, мышцы истончаются, теряется возможность самостоятельно передвигаться. Нарушения дыхания не происходит, поэтому продолжительность жизни таких больных не отличается от показателя здоровых людей.
Диагностика
Врачу важно тщательно собрать анамнез пациента, проанализировать его жалобы. Важно провести неврологический осмотр больного, чтобы установить, атрофированы ли мышцы, нет ли нарушения глотания, мышечной слабости и тонуса, оценить распространенность патологического процесса.
Проводится генетическое консультирование и исследование, электронейромиография для оценки нарушений двигательных нервных волокон, рентгенография длинных трубчатых костей и позвоночного столба.
Также важно оценить изменения общего анализа крови и уровень креатинфосфокиназы в биохимии крови. Уровень последней увеличивается при данных патологиях.
Лечение
Симптомы и лечение спинальной мышечной атрофии у детей и взрослых неразрывно друг с другом связаны. В зависимости от клинической картины заболевания врач определяет тактику лечения и перечень необходимых процедур.
Больным важно понимать, что патология неизлечима, и все методики направлены на устранение ее прогрессирования и укрепление организма. Недавно был зарегистрирован препарат «,Спинраза»,, но используется он пока только на территории США.
Лекарственные средства не способны избавить пациента от заболевания, но могут улучшить общее состояние, препятствовать прогрессированию патологии. Препараты принимаются курсами минимум дважды в год, после осмотра и назначения врача.
Пациентам атрофией мышц назначают следующие средства:
- Витамины группы В, как в таблетированной, так и в инъекционной форме. Выбор средств большой – «,Мильгамма«,, «,Нейромед форте»,, «,Нейромультивит«,, «,Неуробекс»,. Это комплексные препараты. Также используются растворы с содержанием одного вида витаминов. Обычно пациентам рекомендуют пройти курс из 10 инъекций, после чего в течение месяца пьют препараты в таблетированной форме. Схема применения устанавливается врачом.
- Лекарственные средства, улучшающие проводимость нервного импульса. Они также применяются как в виде инъекций, так и в виде таблеток. Препараты данной группы – «,Нейромидин«,, «,Ипигрикс»,, «,Прозерин»,. В начале приема больным вводят средства внутримышечно, курс – 10 инъекций. После этого препараты принимаются внутрь в течение 1-1,5 месяца.
- Лекарственные средства, влияющие на обмен веществ тканей центральной и периферической нервной системы, – «,Цитофлавин»,, «,Церебромедин»,, «,Церебролизат»,, «,Карнитин»,, антиоксиданты и препараты с аминокислотами. Они вводятся курсами внутривенно и внутримышечно в среднем 10 раз.
- Ноотропы и нейропротекторы – гамма-аминомасляная кислота, «,Цитиколин»,, «,Мексидол»,, «,Сермион», и другие. Данные препараты улучшают функционирование центральной нервной системы, схема приема зависит от конкретного средства.
Также таким пациентам может понадобиться прием антидепрессантов. Их назначением занимается психотерапевт, и выбор средства осуществляется индивидуально – обычно предпочтение отдается таким препаратам, как «,Сертралин»,, «,Грандаксин»,, «,Сульпирид»,.
Больным рекомендовано исключить из рациона жирные, жареные, соленые, острые блюда, консервы и пряности. Меню должно включать в себя овощи, фрукты, зелень, нежирные сорта мяса и рыбы. Следует ограничить себя в сладостях. Также исключаются спиртные напитки и газировки.
Питание должно быть дробным, 4-5 раз в сутки, маленькими порциями. Сама еда при этом не должна быть ни горячей, ни холодной. Также следует отметить, что больным рекомендовано выпивать по 0,5 литра чистой воды за 10 минут до основного приема пищи.
Пациентам с синдромом СМА показано посещение сеансов физиотерапии. Им назначают лечебные ванны, электростимулирующие процедуры. В среднем требуется 10 сеансов, которые больные проходят курсами.

Массаж при спинальной мышечной атрофии полезен, помогает держать мышечный корсет корпуса и конечностей в необходимом тонусе, препятствует его истончению. Процедуру необходимо проходить курсами, минимум дважды в год, только в медицинском учреждении, где ее будет осуществлять обученный специалист.
Больным с данным диагнозом важно ежедневно делать упражнения лечебной физкультуры, которые помогут укрепить мышцы и будут препятствовать их атрофии. Полезно заниматься плаванием, скандинавской ходьбой. Также важно делать упражнения для укрепления мышц спины и конечностей.
Особое значение придается дыхательной гимнастике (самый простой пример – надувание воздушных шариков), которая предупреждает развитие застойных процессов в грудной клетке.
Обучение пациентов осуществляется опытным медицинском персоналом, который должен проследить за правильностью выполнения всех упражнений, ведь в дальнейшем больной их будет делать в домашних условиях.
Узнайте, как укрепить мышцы спины.
Методы народной медицины не используются при лечении спинальной мышечной атрофии. Заболевания данной группы —, наследственные, и подобное лечение не способно повлиять на их причины, течение и прогрессирование. Об этом важно помнить всем пациентам с данным диагнозом.
Профилактика
Каких-либо мер профилактики этих заболеваний не существует. В период беременности важно проходить консультацию генетика с необходимыми обследованиями, которые могут подтвердить или опровергнуть неблагоприятный диагноз. Врач может предложить прервать беременность при плохом прогнозе, но решение всегда принимается родителями.
Заключение
Важно иметь информацию о том, что это – диагноз СМА, знать его причины, методы лечения. Это поможет своевременно заподозрить патологию и посетить врача. Правильно поставленный диагноз способствует вовремя начатой терапии, что важно для того, чтобы избежать осложнений и увеличить продолжительность жизни пациента.
Читайте также:
