
Реабилитация при спинальной мышечной атрофии
Спинальная мышечная атрофия или СМА – генетически обусловленная патология, обнаруживаемая у младенцев, детей дошкольного возраста, подростков и взрослых и сопровождающаяся равносторонней атрофией нейронов спинномозговых передних рогов и корешков периферических нервов, что приводит к снижению мышечного тонуса и прогрессирующему параличу. В первом случае медики вынуждены констатировать тот факт, что ребенок никогда не сможет самостоятельно стоять, сидеть и ходить. В остальных он будет постепенно утрачивать эти способности и однажды окажется прикованным к инвалидному креслу.


Что такое спинальная мышечная атрофия и ее виды
Под этим термином объединяется несколько различных видов наследственных заболеваний, сопровождающихся ограничением двигательных способностей. Этим и объясняется тот факт, что в части случаев нарушения обнаруживаются не в младенческом возрасте, а у подростков или уже зрелых людей.
Впервые заболевание было описано в 1891 г. Г. Верднигом и в 1892 г. было выделено в отдельную нозологическую единицу Дж. Хоффманом, благодаря стараниям которых и получила свое второе название. Примерно через полвека Е. Кугелбергом и Л. Веландером была открыта другая подобная болезнь, развивающаяся в более позднем возрасте и отличающаяся более благоприятным течением.
Различают следующие формы патологии:
- СМА 0;
- СМА 1 (тяжелая форма);
- СМА 2 (промежуточная форма);
- СМА 3 (легкая форма);
- СМА 4 (поздняя форма).
Все их объединяет то, что причина их возникновения кроется в мутации рецессивного гена 5 хромосомы SMN. Это приводит к сбоям в продукции протеинов в организме, являющихся строительным материалом всех клеток. В результате страдают мотонейроны спинного мозга и постепенно разрушаются. Поскольку без них невозможна передача нервных импульсов к мышечным волокнам, они постепенно атрофируются, что становится причиной утраты способности двигаться.

К счастью, даже при наличии у обоих родителей мутации гена SMNу них с 75% вероятностью может родиться здоровый ребенок. Но практически всегда он также будет носителем этого гена. Поэтому при планировании беременности стоит проходить генетическое исследование, особенно при наличии случаев СМА в семье.
Это врожденная болезнь, признаки которой обнаруживаются обычно еще в роддоме. Она встречается редко и ее часто объединяются со СМА-1. Для этого вида типично абсолютное отсутствие подвижности, слабость мышц, отсутствие сухожильных рефлексов и ограничение функциональности коленных суставов. С первых дней жизни ребенок страдает от нарушения дыхания.
Спинально-мышечную атрофию важно дифференцировать с перинатальной энцефалопатией и родовыми травмами, но если при них состояние детей постепенно улучшается, то при СМА оно не меняется. Более того часто присоединяются осложнения, которые практически всегда приводят к смерти младенцев в течение первого месяца жизни.

Этот тип течения спинальной мышечной атрофии характеризуется очень тяжелым протеканием. Обычно она обнаруживается до 6-ти месяцев и сопровождается слабостью мышц, периодическими спазмами, что сложно заметить в связи с особенностями анатомии детей первого года жизни (присутствия ярко-выраженной подкожно-жировой клетчатки).
Также заболевание проявляется регулярно пробегающей по языку дрожью, снижением рвотного, сосательного, глотательного рефлексов. Это приводит к возникновению серьезных трудностей при кормлении. Присутствует нарушение слюноотделения, кашель. Ребенок часто громко кричит.
Эта форма спинально-мышечной атрофии может сопровождаться олигофренией и врожденными пороками сердца. Дети подвержены тяжелым нарушениям дыхания, развитию воспаления легких. В связи с этим более половины детей не доживает до 2 лет и только 10% могут отметить свой 5-летний юбилей. Причиной смерти становятся пневмония, остановка сердца или дыхательная недостаточность.
Заболевание обнаруживается у детей от 6 месяцев до 1,5–2 лет. Поэтому такую форму СМА часто называют поздней младенческой. Для нее типично:
- слабость и дрожь в мышцах;
- тремор пальцев, языка;
- скованность движений, обусловленная ограничением подвижности конечностей;
- задержка развития;
- недобор веса.
Дети с таким диагнозом способны самостоятельно сидеть, играть, есть, но стоять и передвигаться нет. К сожалению, патология склонна прогрессировать, что приводит к постепенному ослаблению мышц груди и шеи, следствием чего становится невозможность удерживать голову прямо и часто она безвольно свисает. Затем пропадают сухожильные рефлексы, слабеет голос и отмечаются нарушения акта глотания.
Длительность жизни при таком диагнозе составляет около 10–12 лет. Но треть больных погибает в возрасте до 4-х лет.
Спинальную мышечную атрофию этого вида диагностируют обычно после 2 лет. Она так же проявляется слабостью мышц, но не в такой степени как при СМА 1 или даже СМА 2. Больные могут самостоятельно стоять, но только в течение короткого периода времени. В связи с атрофией мышц это дается им с трудом.
Несмотря на имеющееся заболевание, до 10–12 лет ребенок развивается нормально, что может ввести его родных в заблуждение и вызвать сомнения в правильности поставленного диагноза. Но, достигая этого временного рубежа, возникают первые признаки СМА. Ребенок начинает спотыкаться чаще обычного, падает и не может выполнять физическую работу или заниматься спортом, часто сталкивается с переломами. Постепенно бег, а затем и ходьба даются все сложнее из-за возникновения ограничения подвижности суставов. Впоследствии подросток теряет способность передвигаться без инвалидного кресла.
Прогрессирование патологии приводит к возникновению тяжелого сколиоза, что влечет за собой изменение формы грудной клетки и появление трудностей при дыхании. Именно в этом таится главная угроза болезни для жизни.
К этому типу заболевания относят несколько разных не влияющих на продолжительность жизни, но приводящих к инвалидизации амиотрофий:
- бульбоспинальную Кеннеди;
- дистальную Дюшена-Арана;
- перонеальную Вюльпиана.
Их объединяет то, что первые клинические признаки заболевания проявляются в период от 16 до 60 лет, чаще в 35–40 лет. Это сопровождается угасанием сухожильных рефлексов и заметными спазмами мышц. При атрофии Дюшена-Арана сильнее всего страдают кисти, а для болезни Вюльпиана характерно изменение формы лопаток на крыловидную.
Симптомы СМА
Различные разновидности заболевания объединяет череда общих проявлений, хотя каждая из них имеет и специфичные симптомы. К числу общих признаков принадлежат:
- нарастающая мышечная слабость и постепенная атрофия;
- при тех видах спинальной мышечной атрофии, что обнаруживаются после года или двух лет, наблюдается деградация имеющихся физических достижений, к примеру, способности бегать, ходить;
- тремор пальцев, языка;
- искривление позвоночника;
- частое сохранение нормального психического развития и умственных способностей.
Статистика показывает, что чаще СМА поражает мальчиков.
Диагностика
Предельно информативным методом диагностики СМА считается генетический анализ. Его можно проводить ребенку и взрослому в любом возрасте, а с целью ранней диагностики его выполнение возможно еще на этапе внутриутробного развития. При невозможности проведения анализа ДНК и для окончательного подтверждения диагноза назначаются:
- биохимический анализ крови;
- гистологический анализ мышечных волокон;
- МРТ;
- электромиография;
- микроскопия спинного мозга;
- тандемная масс-спектрометрия.

Лечение спинально-мышечная атрофия
Больным назначается комплексная консервативная терапия, направленная на улучшение способности нервных импульсов проходить к мышцам и работы головного мозга. В этих целях рекомендуется прием:
- ноотропов;
- препаратов α-липоевой кислоты, ацетил-L-карнитина, α-глицерофосфохолина;
- витаминных комплексов, включающих, прежде всего, витамины группы В;
- средств, улучшающих обмен веществ.

Сегодня в разработке находятся специфические лекарственные средства, способные воздействовать на причину развития СМА – дефицит ряда белков. Но в данный момент они находятся на стадии испытаний. Пока что единственным способом хотя бы частично обеспечить организм необходимыми белками является соблюдение специальной диеты. Она подразумевает употребление продуктов, богатых на аминокислоты, а именно зерновых культур, орехов, кисломолочных продуктов, рыбы, мяса. Весьма полезно включение в меню шпината, брокколи, грейпфрутов. Особенно ценны блюда из бурого риса и овса.
Для поддержания мышечного тонуса рекомендованы:
- занятия ЛФК;
- массаж;
- физиотерапевтическое лечение;
- нейромышечная стимуляция.
Современная медицина способна помочь пациентам с СМА за счет выравнивания позвоночника. Вы можете существенно повысить качество жизни и избавиться от болей с помощью хирургического лечения нейромышечного сколиоза. Наши спинальные хирурги способны грамотно провести операцию с учетом всех особенностей пациента и добиться предельно высоких результатов. Цены наших услуг приведены в прайсе.
Суть хирургического лечения нейромышечного сколиоза заключается в выполнении многоуровневой фиксации позвоночника с помощью специальных конструкций. Это предполагает изменение и закрепление в максимально приближенном к нормальному положению каждого сегмента искривленной части позвоночного столба.
Многоуровневая фиксация реализуется за счет установки многочисленных опорных элементов и выбора в качестве опорных точек крестца и таза и позвонков верхнегрудного отдела. Но часто ее проведение требуется практически по всей длине позвоночника, так как у больных спинальной мышечной атрофией сколиотические деформации достигают предельно тяжелых форм.
Она позволяет не только практически полностью выровнять позвоночник, но и равномерно распределить нагрузку на него, а также надежно удерживать его в новом положении. Благодаря этому больной избавляется от выраженного комфорта во время сидения и лежания, решаются психологические проблемы, спровоцированные выраженной деформацией позвоночного столба. Но главное достоинство операции заключается в устранении негативного влияния сколиоза на легкие и другие внутренние органы.
Стоимость коррекции сколиоза при СМА от 640 000 руб и зависит от:
— Тяжести заболевания (сколько времени пациент проведен в стационаре после операции)
— Фирмы производителя имплантов;
— Клиники (где будет проведена операция) и класса палаты.
Цена включает в себя:
— Прибывание в клинике до и после операции;
— Импланты.
— Операцию;
— Наркоз;
— Нейрофизиологический мониторинг
— Наблюдение и консультация на период реабилитации.
Все услуги клиники и стоимость приведены в прайсе.
На первичной консультации ответим на все интересующие вас вопросы, точно определим возможные риски и потенциальную пользу хирургического лечения и подарим вашему ребенку если не возможность ходить, то уверенно сидеть без болей и психологического дискомфорта.


Кто ответит
После пикета советник вице-губернатора Красноярского края Андрей Агафонов пристыдил отца двух тяжелобольных детей в своем telegram-канале.

Никита Рукосуев на одиночном пикете

Дорогой укол
Для реализации этого законопроекта потребуется выделить приблизительно 48 млрд рублей в 2021 году, 29 млрд — в 2022-м и более 32 млрд — в 2023 году.
СМА считают одним из самых частых среди редких (орфанных) заболеваний. Оно встречается у одного ребенка из 6–10 тыс. Болезнь поражает двигательный нейроны спинного мозга, приводит к атрофии мышц и органов дыхания.
В настоящий момент спинраза — единственный зарегистрированный в России препарат для лечения СМА. Стоимость каждой инъекции составляет около $125 тыс. Спинраза назначается курсовым лечением, и уколы делают непосредственно в спинной мозг пациента. В первый год терапии необходимо как минимум шесть инъекций, а дальше уколы делают три раза в год в течение всей жизни.

В перспективе авторы законопроекта полагают, что в России будет зарегистрирован инновационный препарат золгенсма. Всего одна инъекция этого препарата способна остановить развитие болезни. Его стоимость составляет порядка 150 млн рублей, но он подходит не всем. В случае если укол успеют сделать ребенку до достижения двухлетнего возраста, болезнь может полностью отступить. Если же лечение начали после двух лет, полностью восстановиться двигательные нейроны уже не получится.
По его словам, в текущих обстоятельствах необходимо принимать законы прямого действия, а не рамочные законопроекты.
Появился шанс

— Пациенты начали сталкиваться с отказами региональных властей в оплате лечения. Эта ситуация затрагивает несколько сотен российских семей. Вокруг детей со СМА в России объединяется огромное число людей. Это и сами семьи, их друзья, знакомые, неравнодушные люди. Подключаются депутаты и сенаторы. Мы регулярно получаем запросы от региональных депутатов с просьбой помочь оплатить лечение для конкретного ребенка, но пациентская организация не обладает такими ресурсами. Это должно осуществляться за счет государства. Очень хорошо, что появляются депутаты, которые понимают, что в их власти решать вопросы по-другому, — пояснила Германенко.
Суть законопроекта заключается не в том, чтобы включить спинразу в перечень жизненно необходимых лекарств, а в том, чтобы включить само заболевание в программу затратных нозологий. Это означает, что любые зарегистрированные препараты для этих пациентов могут финансироваться за счет бюджета. На сегодняшний момент спинраза — единственный зарегистрированный препарат, поэтому посчитать затраты можно только на его примере.

— Ряд регионов начал выходить на аукционы для единичных пациентов. Мы говорим буквально о двух десятках детей, которые либо уже получили лекарство, либо его получат, но есть еще сотни детей, которые не могут его добиться. Надо снимать нагрузку с регионов: они отказывают просто потому, что не тянут финансово, — добавила Германенко.
— Решение одного лекарственного вопроса не решит полностью проблему СМА. Нам нужно будет работать над тем, чтобы медицинские организации по всей стране формировали свои центры компетенции по работе с этой нозологией, потому что она одна самых распространенных среди редких. Теперь мы знаем, что нам можно не только симптоматически помогать — то есть снимать симптомы проявления болезни, но и помогать радикально с помощью современной терапии, — резюмировала Германенко.
Болезнь не знают

В 2020 году уже умерли трое пациентов, не дождавшихся жизненно необходимого лекарства. В ноябре прошлого года из-за этой болезни скончались еще семеро детей. В федеральном бюджете на 2020 год нет расходов на лечение пациентов со СМА, эти средства выделяют регионы. Однако лекарства закупают в единичных случаях, чаще всего врачи отказываются давать назначение на дорогостоящий препарат.
Родители 10-месячной Вари Ростовой из Ставропольского края собирают деньги на препарат золгенсма уже несколько месяцев. Стараниями неравнодушных удалось собрать большую, но всё еще недостаточную сумму — 9,8 млн рублей из необходимых 150 млн. Каждый день состояние ребенка ухудшается: девочка теряет двигательные навыки. При этом интеллект детей со СМА остается полностью сохранным. Четыре месяца врачи не могли поставить диагноз: узнать его удалось только после платного генетического исследования.
— В провинции об этой болезни никто не знает, даже в краевой больнице мало специалистов, которые слышали о ней. Это одна из главных проблем. В октябре мы впервые услышали диагноз СМА. Но никто из врачей не смог ничего о нем рассказать, посоветовали почитать в интернете. Информации о том, куда обратиться и что делать, нет фактически нигде. Обо всем мы узнавали из чата таких же родителей и подали документы в единственный фонд, который занимается пациентами со СМА, — рассказал отец девочки Станислав Ростов.
Препарат золгенсма стоимостью более $2 млн
После этого родители писали заявки в Минздрав. Но получить назначение на этот препарат оказалось фактически невозможно.

— Полгода наш ребенок фактически был без лечения, а для таких детей это критический срок. Врачи слышат диагноз и чуть ли не крестятся. Может, у них какое-то негласное соглашение, но добиться назначения нам не удалось. Мы подали заявку в Национальный центр здоровья детей и получили ответ, что всё в руках региональной больницы. То есть места, где не могут даже поставить диагноз. Обратились в клинику в Израиле и прислали счет в размере 47 млн рублей на первый год лечения. Дальше каждый год нужно платить 22 млн до конца жизни, — добавил папа Вари Ростовой.
Практически все страны Европы начиная от Македонии и Польши и заканчивая Испанией и Великобританией обеспечивают своих пациентов со СМА бесплатным лечением. Люди с неизлечимым диагнозом получают спинразу вне зависимости от формы болезни и возраста больного.
Первой в мире инновационный препарат золгенсма получила Катя Рубцова из Москвы. Ее родители также открыли сбор на лечение и им удалось отправиться в американскую клинику благодаря крупному пожертвованию анонимного мецената. Ребенку ввели препарат за несколько дней до двухлетия и добились поразительных результатов. В России сбор на этот препарат закрыли уже три семьи, и еще ни разу родителям не удалось собрать необходимую сумму в столь короткий срок без вмешательства крупных жертвователей.
Фонды отказывают

Швейцарская фармацевтическая компания Novartis — производитель золгенсма устроила лотерею, в которой больные дети могут получить бесплатное лечение. Но чтобы участвовать в ней, нужно оформить заявку на английском языке, и делать это должен специалист, который согласится везти ребенка на лечение.
Жители города Георгиевска вышли на сход в поддержку Вари. Семье помогает команда волонтеров, которые выходят на городскую площадь с плакатами и рассказывают горожанам о девочке со СМА. Местные фирмы помогают печатать листовки и наклейки. Волонтеры организовали благотворительный концерт в городском доме культуры, где выступили местные вокалисты и танцевальные коллективы.
— В нашем городе 70 тыс. человек, и тут буквально каждый знает про Варю. 150 млн рублей в масштабах страны — это песчинка, а в масштабах одной семьи — неподъемно. Сейчас нам помогает мэр, очень рассчитываем, что на проблему обратит внимание губернатор [Ставропольского края Владимир Владимиров. — Прим. ред], — добавил отец девочки со СМА.
У одного из 6-10 тысяч детей есть мутация в гене, которая со временем не даст ему двигаться, есть и даже дышать. Но ребенок может вести нормальную жизнь, если вовремя получит терапию, желательно до того, как появятся первые признаки спинальной мышечной атрофии (СМА).
Первое лекарство для пациентов со СМА появилось всего несколько лет назад и доходит до российских пациентов с трудом. Сведения о новых средствах одновременно противоречивы и полны надежды.

Фото: Sebastian Gollnow/dpa/picture-alliance
Что такое СМА
Спинальная мышечная атрофия (СМА) — это неизлечимое заболевание, при котором у человека поврежден или вовсе отсутствует ген, отвечающий за работу двигательных нейронов. Болезнь приводит к поражению нервной системы и постепенной атрофии мышц. В результате у человека сильно искривляется позвоночник, ему становится трудно дышать, он не может двигаться, при этом его интеллект полностью сохранен.
СМА может проявиться как в первые месяцы жизни, так и в возрасте от полутора до двух лет. Но чем раньше диагностировать заболевание, тем лучше для ребенка, — лечение наиболее эффективно, когда симптомы еще не проявились. Поэтому эксперты настаивают на том, что всем новорожденным нужно проводить скрининг на СМА. Болезнь неизлечима, но если человек вовремя получает лекарства и специальное медицинское оборудование, то может полноценно жить.
Спинраза
Рисдиплам
Золгенсма
Однако эксперты относятся к результатам с осторожностью: наблюдения за участниками испытаний длятся не больше четыре лет. Кроме того, в тестированиях принимали участие только несколько десятков детей со СМА в возрасте около семи месяцев, тогда как компания рекомендует использовать препарат детям до двух лет.
Крик отчаяния
Закрыть сбор на препарат для Кати помог анонимный благотворитель, который пожертвовал на лечение девочки 145 миллионов рублей. Сейчас надеются собрать средства на лечение семьи из Зеленограда, Калининграда, Мурманска, Воронежа, Москвы, Красноярска, Екатеринбурга, Владикавказа, Югорска и Воронежа.
Каждый день мы пишем о самых важных проблемах в нашей стране. Мы уверены, что их можно преодолеть, только рассказывая о том, что происходит на самом деле. Поэтому мы посылаем корреспондентов в командировки, публикуем репортажи и интервью, фотоистории и экспертные мнения. Мы собираем деньги для множества фондов — и не берем из них никакого процента на свою работу.
Пожалуйста, подпишитесь на любое пожертвование в нашу пользу. Спасибо.

На Ваш почтовый ящик отправлено сообщение, содержащее ссылку для подтверждения правильности адреса. Пожалуйста, перейдите по ссылке для завершения подписки.
Исключительные права на фото- и иные материалы принадлежат авторам. Любое размещение материалов на сторонних ресурсах необходимо согласовывать с правообладателями.
По всем вопросам обращайтесь на mne@nuzhnapomosh.ru
Нашли опечатку? Выделите слово и нажмите Ctrl+Enter
- О фонде
- Контакты
- Отчеты
- Для НКО
- Персональные данные
- Пожертвовать
- Стать волонтером
- Частые вопросы
- ВКонтакте
- Telegram
- Youtube
- Дзен
Нашли опечатку? Выделите слово и нажмите Ctrl+Enter
(Протокол № 1 от 20.01.2020 г.)
Благотворительный фонд помощи социально-незащищенным гражданам "Нужна помощь"
Адрес: 119270, г. Москва, Лужнецкая набережная, д. 2/4, стр. 16, помещение 405
ИНН: 9710001171
КПП: 770401001
ОГРН: 1157700014053
Номер счета получателя платежа: 40703810238000002575
Номер корр. счета банка получателя платежа: 30101810400000000225
Наименование банка получателя платежа: ПАО СБЕРБАНК РОССИИ г. Москва
БИК: 044525225
Персональные данные обрабатываются Фондом для целей исполнения договора пожертвования, заключенного между Вами и Фондом, для целей направления Вам информационных сообщений в виде рассылки по электронной почте, СМС-сообщений. В том числе (но не ограничиваясь) Фонд может направлять Вам уведомления о пожертвованиях, новости и отчеты о работе Фонда. Также Персональные данные могут обрабатываться для целей корректной работы Личного кабинета пользователя Сайта по адресу my.nuzhnapomosh.ru.
Персональные данные будут обрабатываться Фондом путем сбора Персональных данных, их записи, систематизации, накопления, хранения, уточнения (обновления, изменения), извлечения, использования, удаления и уничтожения (как с использованием средств автоматизации, так и без их использования).
Передача Персональных данных третьим лицам может быть осуществлена исключительно по основаниям, предусмотренным законодательством Российской Федерации.
Персональные данные будут обрабатываться Фондом до достижения цели обработки, указанной выше, а после будут обезличены или уничтожены, как того требует применимое законодательство Российской Федерации.
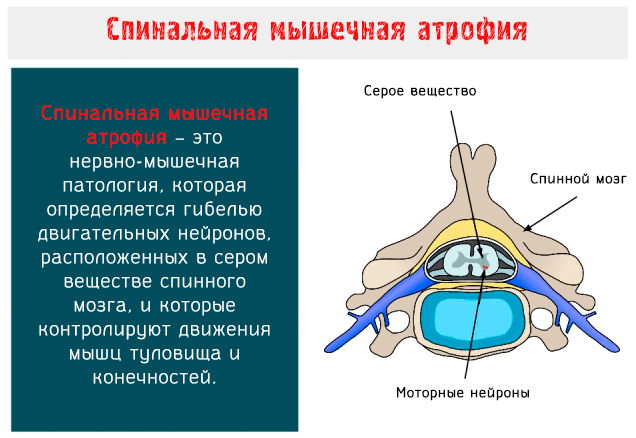
Спинальная мышечная атрофия является основной генетической причиной смерти в детском возрасте. Давайте разберемся в причинах и способах изменения жизни ребенка, а также узнаем, какие методы лечения доступны на сегодняшний день.
Что такое спинальная мышечная атрофия
Спинальная мышечная атрофия (SMA: Spinal Muscular Atrophy) – это нервно-мышечное заболевание аутосомно-рецессивного типа, характеризуется гибелью двигательных нейронов, расположенных в переднем роге серого вещества спинного мозга и в нижней части ствола головного мозга.
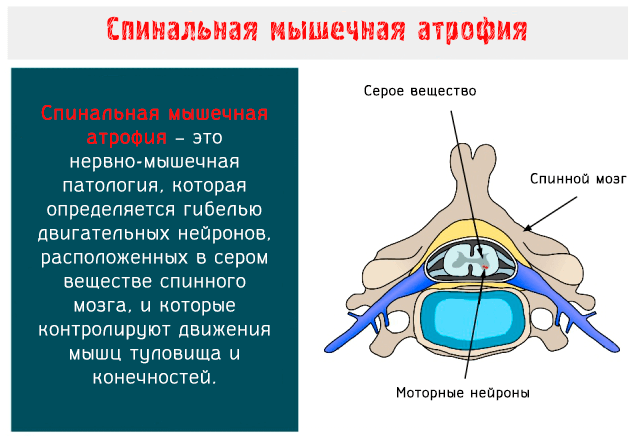
Моторные нейроны – это клетки, из которых образуются нервы, предназначенные для управления скелетными и поперечно-полосатыми мышцами глотки и гортани: когда они вырождаются, целые группы волокон подвергаются атрофии и, соответственно, результатом является мышечная слабость.
Работы глазных мышц, хотя и управляется моторными нейронами энцефального ствола мозга, не нарушается при этой болезни.
Частота спинальной мышечной атрофии колеблется от 1:6000 до 1:10000, и подвержены ей все этнические группы; является редким заболеванием, является одним из самых распространенных нервно-мышечных заболеваний, точнее вторым после дистрофии Дюшенна.
Причина спинально мышечной атрофии
Причина спинально мышечной атрофии была обнаружена в середине 90-х годов, спустя сто лет после первого описания болезни. В 95% случаев речь идёт о делеции в гене SMN1, локализованном на длинном плече хромосомы 5 (делеция – это потеря последовательности ДНК).
Поскольку спинально мышечная атрофия наследуется по аутосомно-рецессивному типу, для развития болезни человек должен получить обе копии плохого SMN1 – от матери и от отца. Таких родителей называют гетерозиготными или носителями, и они не имеют симптомов заболевания. Носители встречаются с частотой 1:50.
Ген SMN1 кодирует белок SMN, который используется в цитоплазме и ядре всех клеток и имеет решающее значение для формирования snRNP, малых ядерных рибонуклеопротеидов, компонентов сплайсинг машин.
Почему же вездесущий белок SMN является критическим фактором для выживания и надлежащего функционирования моторных нейронов?
В 2012 году Лотти и соавторы показали, что белки SMN имеют имеют важное значение для дифференциации и бесперебойной работы моторных нейронов.
Другие гипотезы были сформулированы для объяснения антиапоптической роли SMN:
- потребность в этом белке выше у моторных нейронов, чем в других тканях.
- по мнению других авторов, это можно объяснить тем, что белок SMN участвует в транспортировке вдоль аксонов РНК-связывающих белков.
Несмотря на все предположения, в настоящее время ещё неясно, какая из многих функций протеина SMN связана с развитием спинально мышечной атрофии.
4 типа спинально мышечной атрофии
Спинально мышечную атрофию классифицируют на четыре типа, в соответствии:
- с возрастом появления симптомов
- с максимальной двигательной активностью, на которую способен больной
У 25% лиц избегают точной классификации. Кроме того, у людей, страдающих от одного типа заболевания, симптомы могут существенно различаться.
Это самая тяжелая форма спинально мышечной атрофии, составляет 50% от всех случаев.
Главные её особенности:
- проявляется до 6-го месяца жизни
- ребенок имеет плохую и дряблую мышечную массу: он мало движется, потому что не может противостоять силе тяжести, не в состоянии держать голову в вертикальном положении и сидеть без поддержки
- кости хрупкие и подвержены переломам, кроме того, в позвоночнике развивается сколиоз. Проблемы с костями у пациента со спинально мышечной атрофией не удивляют, так как именно физическая активность способствует минерализации костей
- рефлекс сосания и глотания слабый, поэтому такого ребёнка трудно кормить
- грудная клетка ребенка меньше нормы из-за слабости дыхательных мышц. Кашлевый рефлекс слабый, что нарушает процесс избавления от выделений (слизи и твердые частицы, включая микробов)
У детей, страдающих от спинально мышечной атрофии 1 типа, часто развивается пневмония, так как они не в состоянии избавиться от каких-либо патогенных микроорганизмов с кашлем, а также из-за потери контроля глотательных мышц, которые не могут предотвратить попадание слюны и кусочков пищи в легкие. Повторяющие пневмонии ведут, к сожалению, к дыхательной недостаточности.
Для тех, кто страдает от этой формой патологии прогноз неблагоприятный: смерть наступает в течение 2 лет, даже самое хорошее лечение продлевает жизнь только до 5 лет.
Промежуточная форма спинальной мышечной атрофии.
Давайте посмотрим характеристики:
- проявляется между 6 и 18 месяцами
- ребенок показывает задержку в развитии моторики: не в состоянии сидеть, ему нужна поддержка, чтобы стоять, и никогда не научится ходить. Может иметь легкий тремор рук
- при этом типе также отмечается склонность к развитию сколиоза и хрупкости костей
- у некоторых маленьких пациентов дисфагия становится препятствием для поглощения достаточного для развития количества калорий
- кашлевый рефлекс может ослабнуть, облегчая возникновение респираторных инфекций
При спинальной мышечной атрофии 2 типа также высок риск развития дыхательной недостаточности. Прогрессирование симптомов настолько разнообразно, что некоторые пациенты умирают в младенчестве, другие в состоянии достичь зрелости.
Детская форма спинальной мышечной атрофии, которая:
- может возникнуть в возрасте от полутора лет
- по сравнению с предыдущими случаями, дети могут стоять и ходить самостоятельно, эта способность в некоторых случаях сохраняется до зрелого возраста
- наблюдается тремор рук и могут возникнуть проблемы с суставами и сколиоз
- нарушения дыхания и глотания проявляются менее часто, чем при 1 и 2 типе
У людей, страдающих от 3 типа спинальной мышечной атрофии, средняя продолжительность жизни сравнима со здоровыми людьми. Но, из-за проблем с питанием и низкой физической активность, часто имеют избыточный вес.
Продолжительность жизни нормальная.
Как распознать спинальную мышечную атрофию
Специалист по детской неврологии задаст ряд вопросов, чтобы получить подробный отчет о медицинской истории ребенка и его семьи, после процедуры физического обследования, чтобы оценить физическое состояние маленького пациента.
Подтверждение диагноза спинальной мышечной атрофии достигается благодаря генетическому тесту: берут образец крови и исследуют на наличие аномального гена SMN1. Тест можно использовать, чтобы найти носителей.
Поиск неисправного SMN1 также может осуществляться путём биопсии ворсинок хориона, которые являются частью плаценты, что делает возможным пренатальную диагностику в случае:
- если у пары уже был ребёнок, пострадавший от спинальной мышечной атрофии
- партнеры обнаруживают, что являются носителями, но все равно хотя родить ребёнка
Иногда бывает, что нельзя точно утверждать, что это спинальная мышечная атрофия. Тогда используют другие тесты, которые помогают провести дифференциальный диагноз между спинальной атрофией и другими патологиями нервов и мышц:
- электромиография, которая измеряет электрическую активность мышц
- мышечная биопсия, то есть изучение образцов мышечной ткани
- оценка концентрации креатина киназы, фермента уровень которого повышается при повреждении мышц
Как облегчить симптомы спинальной атрофии
На данный момент не существует лекарств для лечения спинальной мышечной атрофии, поэтому пациенты могут воспользоваться только поддерживающим лечением.
- физиотерапия
- диетология
- дыхание
Для пациентов школьного возраста важно, чтобы они активно участвовали в школьных мероприятиях, потому что их физическая инвалидность никак не влияет на способности к обучению.
Физиотерапия необходима независимо от возраста человека. Упражнения позволят максимизировать амплитуду движений, чтобы предотвратить или замедлить потерю мелкой моторики. Дети со спинальной мышечной атрофией 1 и 2 типа получают огромную пользу от гимнастики в бассейне, поскольку вода помогает стимулировать всю мышечную массу.
Пациентам с 3 типом спинальной мышечной атрофии нужны ортопедические устройства (инвалидные коляски, параподы и т.д.), которые обеспечивают удобство и мобильность. Упражнения также важны, поскольку помогают предотвратить сколиоз, который усугубляет проблемы с дыханием и движениями.
Каждый человек, страдающий от спинальной мышечной атрофии, должен иметь свой индивидуальный план питания, чтобы предотвратить последствия недостаточного или избыточного питания.
У тех детей, которые имеют большие трудности при грудном кормлении, пережевывании пищи и глотании, нужно принять меры, чтобы избежать таких осложнений, как аспирационная пневмония.
- Вы можете прибегнуть к использованию назогастрального зонда, который проходит через нос и доставляет пищу в желудок. Его относительно легко установить и снять, но он может протекать, тогда его следует заменить
- Другой вариант – гастростомия, то есть вывод трубки из желудка; является более простым в обслуживании, но процедура выполняется в операционной под наркозом.
Дыхание
Есть в этом случае для со спинальной мышечной атрофией существует три цели:
- пациенты и всех люди, которые вступают в контакт с ними, должны быть вакцинированы, например, против вируса гриппа, пневмококковой инфекции и бактерии коклюша, потому что инфекции дыхательных путей могут быть очень опасными для таких пациентов
- если кашлевый рефлекс слабым, это можно исправить с помощью специального устройства (Cough Assist): оно создает быстрое изменение давления снаружи и внутри легких, и быстрое прохождение воздуха по дыхательным путям, что имитирует кашель, освобождая дыхательные пути от секрета и микробов
- наконец, важна оценка дыхательной функции этих субъектов по степени сатурации кислорода в крови. Если количества кислорода меньше потребностей, стоит серьёзно рассмотреть идею использования механического респиратора. Изначально он используется в случае инфекции дыхательных путей и во время сна; с развитием атрфоии – весь день.
Открытие причины заболевания открыло для исследовательских групп большое направление для поиска методов лечения, направленных на максимально возможное замедление прогрессирования симптомов: повышение уровня белка SMN.
- Поскольку спинальная мышечная атрофия является моногенным заболеванием, это позволяет вмешаться в корень недуга, предоставляя пациентам функционирующий ген SMN1 (генная терапия)
- У лиц, страдающих от спинальной атрофии, но имеющих ген SMN2, можно увеличить экспрессию этого гена и заблокировать исключение экзона 7 во время сплайсинга незрелой мРНК.
В обоих случаях количество функционирующего белка SMN увеличивается.
AVXS-101 – экспериментальный препарат, разработанный биотехнологической компании Авексис, которому удалось достичь 1 этапа экспериментов на людях, при оценке безопасности лечения, и он начинает проверку эффективности.
Авексис сосредоточились на детях, страдающих от спинальной мышечной атрофии 1 типа, потому что это самый распространенный и смертельный тип заболевания.
AVXS-101 состоит из большого числа частиц адено-ассоциированного вируса серотипа 9, неспособного к репликации, но содержащего одну копию нормального гена SMN1.
Вводится в организм внутривенно, в состоянии преодолеть гематоэнцефалический барьер и достичь моторных нейронов.
Молекула ДНК, переносимая каждым вирусным вектором, производится в лаборатории. Она не изменяет ДНК пациента; содержит промотор, т.е. последовательность, которая способствует транскрипции ДНК в РНК, и гарантирует постоянное производство протеина SMN.
Анализ промежуточных данных, опубликованных Авексис в апреле 2016 года показывает, что:
Читайте также:
