
Позвоночник при синдроме марфана
Приветствую, дорогие читатели. Сегодня хотел бы поговорить про высоких людей с Синдромом Марфана, для которых высокий рост – это не просто особенность организма, а симптом серьезной болезни, требующей к себе постоянного внимания.
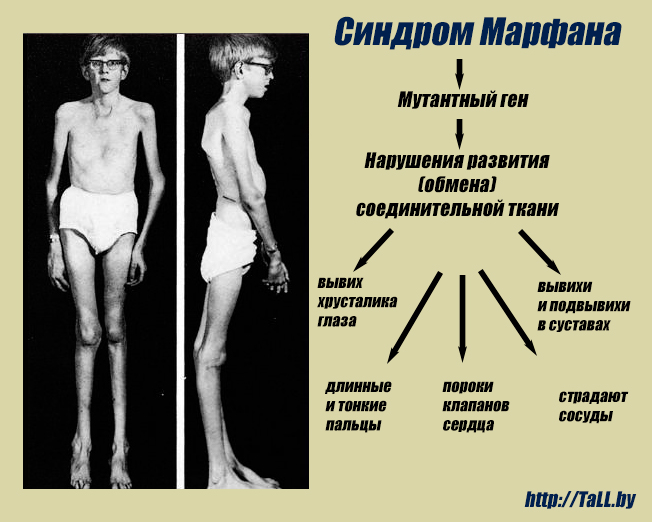
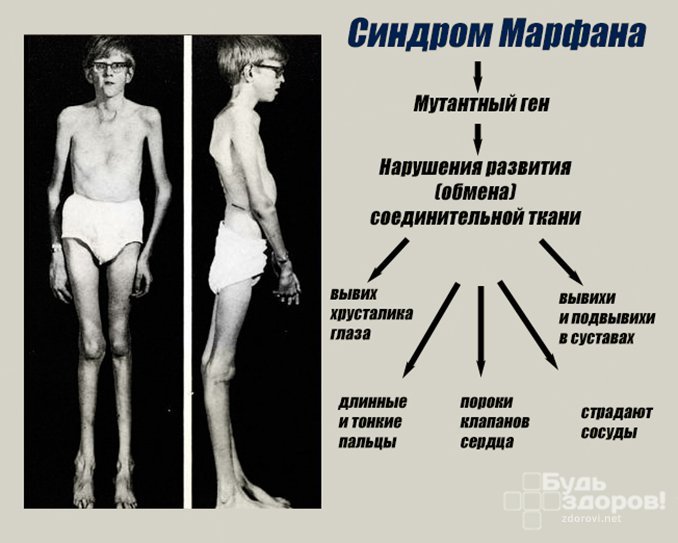
Синдром Марфана — заболевание наследственного типа, при котором поражается соединительная ткань с вовлечением в процесс скелетно-мышечной системы и глаз. Установлено, что причиной патологии является мутация гена фибриллина FBN1. Заболевание полиморфно — может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутации в генах.
Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена.
Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
История заболевания
В 1876 г. симптомы неизвестной патологии были отмечены доктором Вильямсом, но клинические наблюдения проводились гораздо позже — в 1896 г. педиатром из Франции А. Марфаном. Врач в течение 5-ти лет оценивал состояние девочки с неизученными ранее аномалиями, заключающимися в прогрессировании дистрофии скелета и мышечной ткани.
К середине 20-го века имелось множество описанных случаев, когда у больных наблюдались симптомы, близкие к патологии Марфана, и все они относились к заболеваниям наследственного типа. Среди таких случаев — расслоение аорты, пороки сердца, эктопия хрусталиков, сопровождающиеся деформацией костей (грудной клетки, позвоночника) и внешними отклонениями от нормы (высокий рост, худоба, длинные конечности). Американским генетиком МакКьюсиком было проведено детальное исследование мутаций хромосом и открыта новая группа заболеваний соединительной ткани.

Симптомы синдрома Марфана
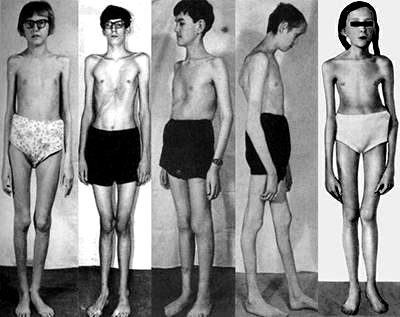
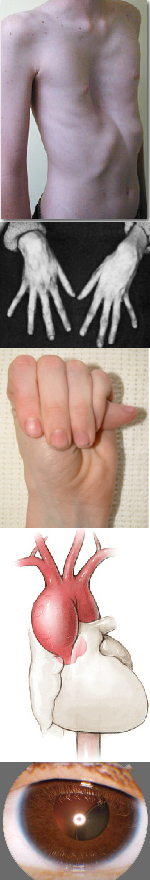
Многообразие вариантов генетической мутации обуславливает различные формы течения болезни. Нередко они малозаметны, иногда приводят к инвалидизации человека в раннем возрасте. Частый признак синдрома Марфана — высокий рост (до 200 см.), при этом туловище непропорционально короткое, а конечности удлиненные и тонкие. Пальцы у больных длинные, паукообразные (арахнодактилия) Из-за недоразвития подкожной клетчатки и мышечной дистрофии страдающие синдромом Марфана имеют астеническое телосложение.
Прочие внешние симптомы патологии (в каждом индивидуальном случае может наблюдаться один или несколько из них):
Более серьезные изменения при синдроме Марфана протекают в организме. Самые тяжелые из них развиваются со стороны сердца и сосудов и могут привести к смерти ребенка еще на первом году жизни. Среди них:
— дефекты ветвей легочной артерии, аорты (расширения, аневризмы, расслоения);
— пороки сердца (чаще — поражения клапанов);
— стенозы артерий.
Подобные нарушения вызывают тахикардию, мерцательную аритмию вплоть до фибрилляции предсердий или развития сердечной недостаточности.
Со стороны глаз наблюдается выраженная миопия, вывих хрусталика, аномалии развития роговицы, уменьшение в размерах радужки, косоглазие, патологии сосудистой стенки сетчатки. При прогрессирующем вывихе хрусталика или при отслойке сетчатки уже в раннем возрасте больные могут полностью потерять зрение.
Со стороны нервной системы при синдроме Марфана происходит растяжение твердой мозговой оболочки и выбухание ликвора в костные дефекты в пояснично-крестцовом отделе позвоночника (дуральная эктазия). Легкие страдают гораздо реже, так как незначительные нарушения их работы не оказывают влияния на дыхательную функцию. Но в отдельных случаях снижение эластичности альвеол может привести к спонтанному пневмотораксу, развитию дыхательной недостаточности. Прочими симптомами патологии могут быть эктопия почек, деформации мочевого пузыря, половых органов.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.
Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения:
— прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.);
— хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Известные люди с синдромом Марфана
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:

Музыкант-скрипач Никколо Паганини. Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.
Писатель Ганс Кристиан Андерсен. Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Писатель Корней Чуковский. Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Источники и ссылки:
1. Причины высокого роста — статья, в которой рассмотрены основные причины высокого роста человека.
2. Синдром Марфана в википедии — о синдроме Марфана в свободной энциклопедии.
3. Русскоговорящее сообщество людей с синдромом Марфана — форум о синдроме Марфана, общение, обсуждение болезни.
4. Международный фонд синдрома Марфана — фонд помощи больным с синдромом Марфана.
5. Группа вконтакте для больных синдромом Марфана.
Понравилась статья? Расскажи друзьям:
Синдромом Марфана называют патологию, характеризующуюся недоразвитием соединительной ткани у детей в эмбриональном или постнатальном периоде, что обусловлено структурными дефектами коллагена и сопровождается поражением преимущественно опорно-двигательного аппарата, сердечно-сосудистой системы и глаз.

Согласно данным разных авторов, частота встречаемости этого заболевания составляет 1 случай на каждые 10-20 тыс. новорожденных, без половой и расовой принадлежности.
Причины патологии
Синдром Марфана – это врожденная аномалия развития, наследуемая по аутосомно-доминантному типу. В основе патологии лежит мутация в гене FBN1, который отвечает за синтез фибриллина – структурного белка межклеточного матрикса, являющегося важнейшим соединением, придающим эластичность и сократимость соединительной ткани.
Таким образом, основной причиной развития синдрома Марфана является дефицит фибриллина, который приводит к нарушению формирования волокнистых структур, потере упругости и прочности соединительной ткани и, как следствие, к невозможности выдерживать физиологические нагрузки.
Примерно в 75% случаев обнаруживается семейный тип наследования этого синдрома, в остальных 25% – первичная мутация.
Согласно исследованиям, чем больше возраст отца (особенно после 35 лет), тем выше риск рождения ребенка с синдромом Марфана.
Классификация заболевания
Синдром Марфана подразделяют на несколько видов в зависимости от количества пораженных у человека систем на:
- Стертую форму, характеризующуюся слабовыраженными изменениями в одной-двух системах;
- Выраженную форму, которая характеризуется изменениями хотя бы в одной системе; патологиями в двух и более системах; слабовыраженными нарушениями в трех системах.
По характеру течения синдром подразделяется на стабильный и прогрессирующий.
Признаки синдрома Марфана
Клинической картине данной патологии свойственны поражения многих и самых разных жизненно важных органов и систем организма: скелета, глаз, нервной и сердечно-сосудистой систем.
В большинстве случаев признаками синдрома Марфана являются:
- Высокий рост;
- Относительно короткое туловище;
- Непропорционально длинные и тонкие конечности (долихостеномелия);
- Удлиненные паукообразные пальцы (арахнодактилия);
- Астеническое телосложение;
- Мышечная гипотония;
- Слаборазвитая подкожная клетчатка;
- Длинный и узкий лицевой скелет (долихоцефалия);
- Высокое аркообразное нёбо;
- Нарушение прикуса.
В среднем длина тела у девочек с синдромом Марфана составляет 52,5 см при рождении, окончательный рост – 175 см, у мальчиков – 53 и 191 см соответственно.
Также этому заболеванию свойственны нарушения функций суставов (гипермобильность), деформация позвоночника (кифоз, вывихи и подвывихи шейного отдела, сколиоз, спондилолистез, кифосколиоз), протрузия вертлужной впадины, плоскостопие.
В клинической картине синдрома доминирует сердечно-сосудистая патология, которая часто и определяет исход заболевания. Проявляется пороками развития перегородок и клапанного аппарата сердца, дефектами структуры стенок сосудов, в частности крупных ветвей легочной артерии и аорты, а также врожденными пороками сердца: стенозом легочной артерии, дефектом межпредсердной и межжелудочковой перегородки, коарктацией аорты и т.д.
Самая неблагоприятная форма синдрома Марфана, проявляющаяся уже при рождении в классическом варианте, приводит к прогрессирующей сердечной недостаточности и, как следствие, к летальному исходу в течение первого года жизни.
В большинстве случаев признаком синдрома Марфана является и патология органа зрения, характеризующаяся близорукостью, косоглазием, гипоплазией цилиарной мышцы и радужной оболочки, увеличением размера и уплощением роговицы, эктопией хрусталика, изменением калибра сосудов сетчатки.
Также у детей с этой патологией диагностируется поражение нервной и бронхолегочной систем и других органов: эктопия почек, разрывы и вывихи связок, варикозное расширение вен, бедренные и паховые грыжи, опущение мочевого пузыря и матки у девочек, поражение кожи и мягких тканей.
Синдрому Марфана характерен высокий выброс адреналина, что является причиной гиперактивности, нервного возбуждения, а иногда умственной одаренности и развития неординарных способностей.
Диагностика синдрома Марфана
Окончательный диагноз может быть поставлен только после комплексного обследования пациента, в котором принимают участие генетик, ортопед, кардиолог и офтальмолог.
Целью генетиков является изучение семейной истории и в том числе выявление близких родственников, которые умерли от сердечно-сосудистых заболеваний. С целью диагностики синдрома Марфана кардиолог назначает эхокардиограмму, ЭКГ, рентген грудной клетки. Офтальмолог обследует глаза с помощью щелевой лампы с целью выявления вывиха хрусталика или каких-либо других патологий. Ортопед обследует грудную клетку и позвоночник на наличие сколиоза, плоскостопия и т.п.
Если подтверждается версия наследственности, то для постановки окончательного диагноза требуется наличие характерных признаков синдрома Марфана как минимум в двух системах организма. В том случае если наследственности нет, то необходимо подтверждение поражения хотя бы трех систем организма.
Лечение синдрома Марфана
Определенных методов лечения синдрома Марфана, к сожалению, не существует. Главная цель терапии – устранение сопутствующих патологий и предупреждение осложнений.
Лечение прогрессирующего сколиоза проводится физиотерапевтическими процедурами и механическим укреплением скелета, в отдельных случаях проводится хирургическая коррекция.
Некоторым больным показана хирургическая пластика аорты, аортального и митрального клапанов.
Коррекция зрения осуществляется с помощью очков или линз, при необходимости назначается лечение катаракты, глаукомы и т.д.
Таким образом, всем пациентам показана симптоматическая терапия.
Марфаноподобный синдром – дифференцированная форма врожденной соединительнотканной недостаточности с наследственным механизмом передачи, характеризующаяся многообразием клиники.
Лечение и наблюдение пациентов осуществляет группа специалистов, основная цель которых замедлить прогрессирование болезни, так как полное излечение не является возможным.
Определение термина

Синдром Марфана — генетическая дисплазия соединительной ткани, проявляющаяся дефектами структур компонентов внеклеточного матрикса (волокон и основного веществ) с последующими морфофункциональными изменениями органов и системы.
Патологические нарушения вызваны в генетическом аппарате в период эмбрионального и постнатального развития, с преимущественным поражением сердечно-сосудистой системы, скелета, органов зрения.
Дисплазия соединительной ткани впервые была описана в 1896 году французским педиатром Антонином Бернардом Марфаном и терапевтом Эмилем Шарлем Ашаром. Поэтому часто синдром Марфана называют синдромом Марфана-Ашара.
Наследственная коллагенопатия относится к сравнительно редким болезням, встречается с частотой 1 случай на 100 тысяч. При синдроме преимущественно поражается восходящая часть аорты, что создает угрозу внезапной смерти. Однако при грамотно организованной терапии и раннем определении дисплазии возможно сохранение качества и продолжительность жизни.
Важно! Беременность при синдроме Марфана опасна, так как существует риск наследования патологии. Также по мере увеличения срока беременности возрастает угроза разрыва аорты, развития инфекционного эндокардита.
Причины синдрома Марфана
Болезненное состояние относится к генным заболеваниям с аутосомно-доминантным типом наследования. В основе патогенеза лежит стойкое изменение генотипа фибриллина, являющегося важным компонентов соединительной ткани и ответственным за синтез, пространственную организацию коллагена.
Разнообразие мутагенеза FBN1 (насчитывает более 1000 видов) объясняет вариабельный фенотипический спектр марфаноподобного синдрома (от незначительных форм проявлений до ярко-выраженных).
Клиническая картина

Фото больных синдромом Марфана описывает человека высокого роста (окончательный рост у мальчиков 190 см, у девочек 175 см), с непропорционально коротким туловищем и удлиненными тонкими конечностями.
Телосложение характеризуется общей худощавостью, шея длинная и тонкая, плечи узкие, лицевой скелет вытянут с изъявлением высокого аркообразного неба и неправильным прикусом.
Симптомы синдрома Марфана могут не определяться или иметь незначительную выраженность при рождении. В течение жизни клинические формы постепенно нарастают.
Универсальных проявлений патологических изменений не существует. Каждый дефект уникальный, и свойственный определенному индивидууму, семье.
Фенотипические проявления условно разделяют на несколько групп, в зависимости от органов и систем, вовлеченных в диспластический процесс:
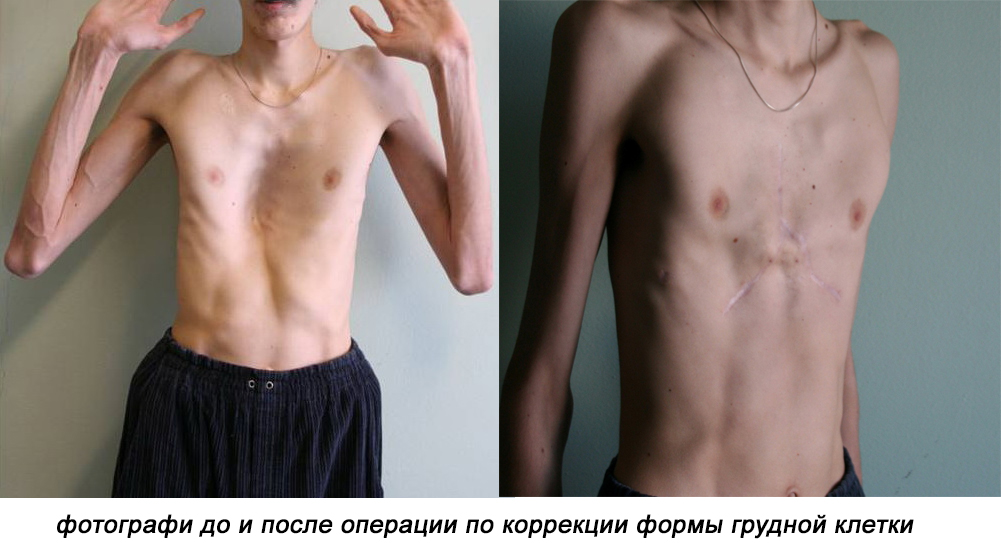
- Костно-суставные изменения: гипермобильность суставов, деформация позвоночного столба (сколиоз, кифоз, спондилолистез), плоскостопие, изменение формы грудной клетки (килевидная или воронкообразная).
- Клапанный и сосудистый синдром: пролапсы клапанов сердца, поражение артерий эластического и мышечного типа, варикозное расширение вен, уплотнение створок митрального клапана, недостаточность митрального кольца, аневризмы, расширение корня аорты.
- Нарушения со стороны органов зрения: миопия, роговица, подвывих хрусталика, гипоплазия радужной оболочки, косоглазие.
Одним из ранних признаков у значительной части пациентов отмечают проявление вегетативной дисфункции. Диагностическими признаками являются диспептические расстройства, боль в ретростернальном пространстве, эпигастрия, тахикардия, сухость слизистых.
В дошкольном и подростковом возрасте манифистирует астенический синдром с жалобами на быструю утомляемость, снижение работоспособности, непереносимость физических и психоэмоциональных нагрузок. Возможен другой исход, на фоне высокого выброса адреналина может развиваться гиперактивность, определяться неординарные умственные способности.
Справка! При синдроме Марфана-Ашара наблюдается поражение и других систем: бронхолегочной (дыхательная недостаточность, спонтанный пневмоторакс), пищеварительной (рефлюкс эзофагит, гастроптоз), мочевыделительной (нефроптоз), кожи и мышц (мышечная гипотония, грыжа).
Методы диагностики синдрома Марфана

Наблюдением и лечением пациентов с наследственной патологией занимается ряд специалистов: терапевт, невролог, эндокринолог, кардиолог, кардиохирург, ортопед, генетик.
Диагностический поиск начинается с изучения семейного анамнеза и анализа клинических критериев. Если в роду была определена дисплазия соединительной ткани, новорожденные подлежат обследованию на генные патологии.
Так как болезнь преимущественно поражает скелет, органы зрения и сердечно-сосудистую систему, то в комплексную программу диагностики необходимо включить следующие мероприятия:
- ЭКГ и ЭхоКГ;
- рентгенографию грудной клетки, тазобедренных суставов;
- МРТ сердца, позвоночника;
- компьютерную томографию;
- офтальмоскопию и биомикроскопию;
- молекулярно-генетический анализ, секвенирование ДНК.
Для определения аутистических черт, сопутствующих расстройств, когнитивных особенностей личности специалисты используют фенотипические диагностические тесты.
Лечение синдрома Марфана
Как лечить синдром Марфана? На сегодняшний день полное излечение этой генной болезни не представляется возможным. Не существует и единой тактики ведения пациентов с наследственной коллагенопатией. Врач должен использовать разные методики, чтобы остановить прогредиентность течения патологии.
Наиболее приемлемым подходом будет коррекция сосудистого, аритмического вегетативного синдрома и прочих нарушений фармакотерапией в сочетании с альтернативной медициной, немедикаментозным воздействием.
Важно! Проблема снижения остроты зрения решается подбором очков или контактных линз. При тяжелых формах может выполняться лазерное или хирургическое лечение катаракты, глаукомы, удаление смещенного хрусталика с заменой на интраокулярную линзу.
Терапевтическая стратегия пациентов с генными дефектами соединительной ткани сводится к коррекции обменных процессов, которые лежат в основе патогенеза данного состояния.
Как фундамент в программе лечения используется комплекс препаратов, принимающих участие в минерализации костной ткани, синтезе и созревании коллагена:
- биостимуляторы;
- витаминные комплексы;
- микро и макроэлементы (марганец, магний, цинк, селен, калий, медь);
- антагонисты кальция.
Особое внимание в медикаментозном лечении синдрома Марфана обращают препараты с метаболической активностью:
Положительные результаты комплексного использования лекарственных средств имеют клиническое обоснование. Специалисты рекомендуют не ограничиваться назначением препаратов кальция и магния, а проводить расширенное метаболическое лечение курсами.
Продолжительность и эпизодичность лечебно-профилактических мероприятий определяется клиническим течением и прогрессированием болезни, общим состоянием пациента.
Какие хондропротекторы помогают при остеохондрозе можно узнать тут.
При выраженных скелетных нарушениях, невозможности восстановления функциональности суставов проводят стабилизирующую операцию на позвоночнике, торакопластическую реконструкцию грудной клетки, частичную или полную замену тазобедренного сустава имплантатом.
Основами реабилитации пациентов с синдромом Марфана с выявлением аритмических и сосудистых нарушений являются ингибиторы ангиотензинпревращающего фермента (АПФ), обладающие гипотензивными свойствами.
Не менее важными базисными медицинскими изделиями в терапии синдрома Марфана будут бета-адреноблокаторы:
Бетаблокаторы обеспечивают восстановление физиологической функциональности сердечно-сосудистой системы, распределение геодинамической нагрузки на аорту, уменьшение выраженности морфологических изменений сосудистой стенки, стабилизацию артериального давления.
Важно! Абсолютными противопоказаниями к назначению β-адреноблокаторов является детский возраст, непереносимость компонентов состава, сердечная недостаточность в острой и декомпенсированной форме, брадикардия, гипотония. Возможны риски связанные с приемом препаратов при беременности и грудном вскармливании.
При значительном расширение аорты (более 5 см), недостаточности сердечных клапанов необходимы реконструктивные и резекционные операции. По отдельным показаниям проводят протезирование митрального клапана.
В реабилитационный период пациент получает антибиотики и антикоагулянты с целью профилактики тромбоза и инфекционного эндокардита.
Медикаментозная терапия полностью не решает проблему дисплазии соединительной ткани, и не может рассматриваться с позиции единственно возможной. Пациенты часто обращаются к альтернативной медицине.

Лечение синдрома Марфана народными средствами не может заменить назначенных доктором ангиопротекторов, бета-адреноблокаторов, метоболических препаратов.
Пациенты с дополняющим подходом получают надежду на достижение большего результата в лечении, продление ремиссии.
Для профилактики обострений и развития осложнений со стороны внутренних органов и систем полезны будут рецепты фитотерапии.
При недостаточности соединительной ткани традиционно используют травы, которые обладают анальгезирующими свойствами, укрепляют венозную систему, улучшают обменные процессы на тканевом уровне.
К таким необходимо отнести:
- одуванчик;
- кору ивы;
- девясил;
- клевер луговой;
- донник;
- сабельник болотный;
- лопух;
- боярышник;
- конский каштан;
- листья березы.
Отвар рекомендован долгим курсом в течение 4-6 месяцев. Схему приема доктор определяет в индивидуальном порядке.
Важно! Методы комплементарной медицины могут оказать серьезное воздействие на организм, как положительное, так и негативное, поэтому выбор вспомогательной лечебной тактики должен обсуждаться с лечащим врачом.
Для коррекции иммунного потенциала, повышения тонуса и защитных сил организма показаны биологические вещества природного происхождения (каменное масло, мумие, экстракт восковой моли), витаминизированные чаи, отвары на основе календулы, солодки, фиалки, череды.
Выраженное значение в стабилизации и поддержании состояния пациента на физиологическом уровне отведено дневному рациону. Задача диетотерапии — восполнить недостаток микро и –макронутриентов, ответственных за синтез коллагена и минерализацию костной ткани.
В меню больных синдромом Марфана включают много овощей, фруктов, зелени, морепродуктов, растительные масла в ассортименте, крупы, печень, красное мясо нежирных сортов, птицу, молочную продукцию
Полностью или частично необходимо исключить из рациона тугоплавкие жиры, концентрированные бульоны, копчености, острые приправы, алкоголь, кофе, кондитерские изделия.
Предотвратить прогрессирование заболевания возможно при условии диагностики и лечения с раннего возраста. Цель профилактических мероприятий – облегчить течение патологии, улучшить качество жизни, минимизировать риски развития тяжелых форм.
Предупреждение синдрома возможно при соблюдении следующих условий:
![]()
Рациональное питание с включением полезных продуктов, восполняющих суточную потребность организма в белках, жирах и углеводах. Особого внимания заслуживают натуральные хондропротекторы, которые принимают участие в строительстве хрящевой и костной ткани, улучшают метаболизм в волокнистом и гиалиновом хряще. Основными источниками хондроитина и глюкозамина является холодец, красная рыба, заливное, желе.- Умеренная физическая нагрузка. Пациентам противопоказаны изометрические упражнения, занятия подводным плаванием, единоборством и другие виды спорта, которые требуют высокой физической активности.
- Постоянное врачебное наблюдение, систематическое выполнение диагностических обследований. Своевременная кардиохирургическая коррекция позволяет увеличить продолжительность жизни пациента до 60-70 лет.

Люди с наследственной коллагенопатией должны избегать стрессовых ситуаций, волнений, психологических перегрузок, как провокаторов соматики.
Заключение
В арсенале врачей находится достаточно широкий спектр препаратов для сдерживания прогрессирования диспластических процессов, предотвращения осложнений. Поэтому главным условием увеличения продолжительности жизни пациента является своевременный диагностический поиск и современная обширная тактика ведения.
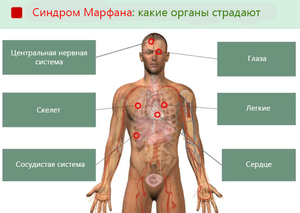
Исследуем симптомы и возможные осложнения, которые могут возникнуть в различных органах из-за такого генетического заболевания, как синдром Марфана.
Давайте посмотрим, как осуществляется диагностика и каковы на сегодняшний день наиболее эффективные методы лечения и увеличения продолжительность жизни.
Что такое синдром Марфана
Синдром Марфана – совокупность симптомов и признаков, составляющих клиническую картину серьезного генетического наследственного заболевания, которое поражает связочные ткани. Точнее, синдром Марфана является следствием мутации гена FBN1, расположенного на хромосоме 15, которая кодирует фибриллин-1, гликопротеин (белок, который содержит полимер, простые сахара или олигосахарид), они имеют важное значение для эластичности волокон соединительной ткани.
Таким образом, синдром Марфана влияет на множество важных органов и тканей: сердце, легкие, скелет, глаза и т.д.
Заболевание передается, как аутосомно-доминантный признак. Большинство больных наследует ген от одного родителей. Замечено, что эти случайные мутации имеют наиболее высокую вероятность проявления у пожилых родителей.
Соединительная ткань выполняет функцию поддержки, объединения и питания тканей организма, которые образуют различные органы. С точки зрения гистологии (наука, которая изучает состав тканей) соединительная ткань может быть различных типов, в зависимости от характеристик ткани и/или органа, которые она образует, но всегда характеризуется наличием клеток, разделенных между собой, но рассеянных в межклеточном веществе как внеклеточный матрикс.
Внеклеточный матрикс состоит из белков, которые образуют волокнистую часть фиброзной ткани и водный раствор белка.
Волокнистая ткань может состоять из трех различных типов волокон: коллагеновых, сетчатых и эластичных. Сетчатые и коллагеновые волокна имеют идентичный химический состав, но отличается структурной организацией. Эластические волокна состоят из двух отдельных белковых цепей в фибриллина и эластина.
Основными видами соединительной ткани являются:
Мы сказали, что Синдром Марфана является следствием мутации гена FBN1, который кодирует белок фибриллин-1. Он необходим для образования эластических волокон, которые составляют структуру соединительной ткани, а также содержат некоторые факторы роста, такие как TGF-beta.
Мутация FBN1 определяет снижение на физиологическом уровне фибриллина 1 и, следовательно, невозможность накопления TGF-beta и ухудшение волокон эластина, то есть возникновение синдрома Марфана.
Симптомы синдрома Марфана – зависят от органов
Учитывая большое количество органов, которые могут быть затронуты заболеванием, симптоматика может быть очень насыщенной (можно обнаружить более 30 различных симптомов и признаков). Кроме того, симптомы синдрома Марфана чрезвычайно изменчивы при переходе от одного лица к другому, даже если они члены одной семьи.

Синдром Марфана поражает.
У некоторых людей они имеют настолько легкую форму, что практически остаются незамеченными для окружающих, у других же симптомы имеют столь острую форму, что это ставит под угрозу жизнь человека.
Так же часто симптомы смешиваются с проявлениями других заболеваний. По этим причинам не всегда диагностика может быть проведена только на основании клинического анализа и в таких случаях будет необходимо прибегнуть к ДНК-тесту.
Возможные симптомы синдрома Марфана сгруппированы на основе влияния на функции различных органов.
- Рост выше нормы, сопровождается чрезмерной худобой.
- Сколиоз (искривление позвоночника).
- Кривые плечи.
- Аномальное искривление грудины.
- Длинные и тонкие верхние и нижние конечности (долихостеномелия), пальцы очень длинные и подвижные, напоминающие лапы паука (арахнодактилия).
- Плоскостопие.
- Очень глубокая вертлужная впадина (полость тазобедренного сустава, в которой расположена головка бедренной кости), что ограничивает движения головки бедренной кости.
- Высокое нёбо и стрельчатая арка, неправильный прикус. Из чего вытекают трудности с произношением звуков.
- Артроз, который развивается аномально рано.
Симптомы сердечно-сосудистой системы:
- Пролапс митрального клапана с астенией, сердцебиением и аритмией.
- Пролапс аортального клапана с недостаточностью аортального возврата крови в левый желудочек.
- Расширение восходящей и брюшной аорты с образованием аневризмы.
- Спонтанный пневмоторакс. Накопление воздуха в плевральной полости. В нормальных условиях давление на внешней поверхности легких меньше, чем атмосферное, и это приводит к тому, что легкие остаются раздутыми. Наличие воздуха в плевральной полости нарушает дыхательную функцию.
- Идиопатическое обструктивное заболевание легких. Болезнь, которая блокирует поток воздуха в легкие и затрудняет дыхание.
- Апноэ сна.
Симптомы центральной нервной системы:
- Дуральная эктазия. Выпячивание в мешочек, который покрывает и защищает спинной мозг. Проявляется болью в спине, головную болью, онемением и слабостью конечностей, невозможностью контроля сфинктеров, а затем недержанием мочи и кала.
- Дегенерация межпозвонковых дисков.
- Дисаутономия. Болезнь, которая влияет на нормальные функции вегетативной нервной системы и, следовательно, контроль частоты сердечных сокращений, кровяное давление и перистальтику (сокращение гладкой мускулатуры органов в виде трубки, таких как кишечник).
- Проблемы рефракции, такие как близорукость и астигматизм.
- Вывих хрусталика. Смещение хрусталика от его обычного положения.
- Отслоение сетчатки.
Диагностика синдрома Марфана: анамнез и показатели
Существует более 200 различных мутаций гена FBN1, молекулярные исследования позволяют определить более 95% случаев генетических дефектов. Но такие анализы могут занять много времени и довольно дорогие, поэтому почти всегда пытаются провести диагностику на основе клиники.
Учитывая большое количество симптомов и многие сопутствующие заболевания, чтобы облегчить задачу врачей, был разработан единый стандарт диагностики. Он предусматривает наблюдение за 7 аппаратами организма, а именно двигательным аппаратом, сердечно-сосудистой системой, легкими, глазами, нервной системой, генетическими маркерами, психикой.
Лечение синдрома Марфана – лекарства и вмешательства
Не существует никакого постоянного лечения синдрома Марфана, поэтому лечение, как правило, заключается в предотвращении и контроле возможных осложнений.
Лекарства при синдроме Марфана используются, в основном, чтобы сохранить под контролем артериальное давления для предотвращения чрезмерного увеличение аорты.
В последние годы используется метод лечения, действие которого основывается на ингибировании TGFβ. Данная терапия так же, главным образом, защищает от расширения аорты.
Также используют различные хирургические методы лечения для исправления некоторых недостатков, типичных для синдрома Марфана, такие как уменьшение сколиоза, исправление аорты, восстановление рефракции глаз.
Осложнения и риски синдрома Марфана

Синдром Марфана приводит к .
По мере того как органы будут затрагиваться в ходе развития синдрома Марфана, могут появиться различные осложнения, в частности:
- Аневризма и рассечение аорты, как восходящей, так и брюшной. Диссекция аорты часто отмечается во время беременности. В этом состоянии сердце матери подвергается чрезмерному напряжению, что может оказаться фатальным для целостности аорты.
- Нарушения митрального и/или аортального клапана. В результате того, что сердце „переутомляется“, что неизбежно ведет к сердечной недостаточности.
- Отслоение сетчатки.
- Вывих хрусталика и его дислокация.
- Возникновение ранней катаракты и глаукомы.
- Деформация скелета (грудина, позвоночник, ноги).
В прежние годы больные с синдромом Марфана редко жили дольше 40 лет. В настоящее время, с развитием методов хирургического вмешательства, пациенты в состоянии спокойно прожить до 70 лет.
Конечно, требуется постоянный мониторинг состояния организма, который позволяет осуществить своевременное вмешательство в случае критических ситуаций.
Наиболее частой причиной смерти является резекция аорты.
Читайте также:
