
Нейрофиброматоз в спинном мозге на мрт

Общие сведения
Код нейрофиброматоз по мкб-10: Q85.0 (нейрофиброматоз незлокачественный). Нейрофиброматоз относится к наиболее распространенной форме наследственной моногенной патологии, частота встречаемости которого варьирует в пределах 1:2000 — 1:40000. Характерен аутосомно-доминантный тип наследования заболевания с частотой проявления (пенетрантностью) приближающейся к 100%. Различий в частоте поражения по половому признаку не выявлено.
В целом по клинико-морфологическим проявлениям выделено и описано восемь типов нейрофиброматоза, наиболее значимыми из которых являются:
- Нейрофиброматоз первого типа (НФ 1). Около 90% всех нейрофиброматозов приходится на тип НФ1, который и является наиболее распространённым среди заболеваний этой группы.
- Нейрофиброматоз второго типа (НФ 2). Встречается значительно реже (1:40000), но имеет более агрессивное течение, чем НФ1.
Некоторые авторы считают, что из восьми форм нейрофиброматоза все, кроме НФ2 считаются абортивными и не должны выделяться в качестве самостоятельных нозологических форм.
Частота встречаемости у новорожденных составляет 1:4000, в русской популяции — 1,28:10 000. Более характерна передача по отцовской линии, чем по материнской. В более половине случаев проявления заболевания минимальны, часто встречаются неполные и моносимптомные формы. Около 80% случаев нейрофиброматоза спорадические и являются результатом новых мутаций. В связи с присущей гену NF1 высокой частоты мутаций, определяющих клиническое развитие нейрофиброматоза, предполагается или чрезвычайно высокая его мутабельность или наличие мутаций в нескольких локусах гена.
Для нейрофиброматоза I типа характерен выраженный полиморфизм клинических проявлений, прогрессирующее течение и частое вовлечение в процесс различных органов и систем с развитием тяжелых осложнений, нередко приводящих к летальному исходу. Болезнь Реклингхаузена (нейрофиброматоз) имеет четко прослеживающуюся тенденцию к медленному прогрессированию. Существует два периода резкого увеличения активности патологического процесса: первый от 5 и до 10 и второй от 35 до 50 лет. При этом, второй период активности в подавляющем числе случаев (около 75%) обусловлен малигнизацией опухолевых новообразований. К провоцирующим факторам, способствующим росту нейрофибром, относятся состояния и возрастные изменения, сопровождающиеся гормональной перестройкой организма: беременность, период полового созревания.
Особенность заболевания является специфическая последовательность манифестации симптоматики нейрофиброматоза 1-го типа в зависимости от возраста пациента, что и обуславливает трудности в его диагностике у детей раннего возраста. Так, нейрофиброматоз у детей первых лет жизни/с рождения может присутствовать только в виде некоторых признаков заболевания, чаще — это крупные пигментные пятна или скелетные дисплазии и плексиформные нейрофибромы, а другие характерные симптомы могут манифестировать значительно позже (к 5-15 годам). А если учесть, что степень их выраженности, течение и скорость прогрессирования патологического процесса варьируют в широких пределах становится понятными затруднения в постановке диагноза, что и способствует его поздней диагностике.
Нейрофиброматоз II типа встречается независимо от НФ-I типа; его проявления определяются локализацией основного патологического процесса и соответствует симптоматике поражения головного/спинного мозга. В спинном мозге чаще в процесс вовлекаются оболочки спинальных корешков шейного и поясничного отделов в виде Шванном (опухоль доброкачественная из шванновских клеток нервных оболочек), конский хвост (задние корешки); при локализации в головном мозге в процесс могут вовлекаться черепно-мозговые нервы, продолговатый мозг и Варолиев мост. Реже в процесс вовлекаются периферические нервы с развитием эпендимом и менингиом.
Средний возраст манифестации признаков заболевания при НФ 2 составляет 20 лет, а на момент постановки диагноза – около 28 лет. Как правило, НФ 1 начинает проявляться в раннем детстве, преимущественно с кожных симптомов, в то время как НФ 2 проявляется в молодом возрасте, преимущественно с развития глухоты, обусловленной развитием вестибулярных шванном и других симптомов, вторичных относительно спинальных шванном и менингиом.
Патогенез
В основе патогенетических изменений нейрофиброматоза 1 типа лежит высокая частота спонтанных мутаций гена NF1 (17q11.2), кодирующего цитоплазматический белок-онкосупрессор нейрофибромин, который вырабатывается в нервных и специализированных клетках нейроглии. Нейрофибромин содержит в своем составе особый домен, который и реализует супрессорный эффект относительно пролиферации клеток преимущественно нейроэктодермального происхождения за счет инактивации адаптерного белка Ras, который является ингибитором структуры митогенных сигнальных путей. При такого рода генетическом дефекте в хромосомах 17q происходит нарушение динамического равновесия регуляции роста и его смещение в сторону пролиферации и образования доброкачественного опухолевого роста.
Описано более 500 видов мутаций в гене на хромосоме 17q, которые и препятствуют осуществлению регулирующей функции гена NF1 в онкогенезе. Механизм развития клинических проявлений до настоящего времени точно неизвестен. Принято считать, что в основе патологии лежит развитие из нервных оболочек различных опухолей (невриномы, нейрофибромы). Примерно половина случаев являются следствием новых мутаций.
Генетический дефект при нейрофиброматозе 2 типа располагается в 22 хромосоме (22q12) и непосредственно кодирует процесс синтеза белка Мерлина, также являющегося супрессором опухолевого роста. Наибольшее значение он имеет в регулировании пролиферации (размножения) клеток нейроэктодермального происхождения. При мутации синтеза Мерлина в одной хромосоме не проявляется на клеточном уровне, а при симметричной мутации (повреждении) аллельного гена в клетке прекращается синтез Мерлина и соответственно, присущее здоровому организму равновесие в регуляции роста смещается (дрейфует) в сторону пролиферации с развитием процесса доброкачественного опухолевого роста.
Классификация
В основе классификации нейрофиброматоза лежат клинико-морфологические особенности заболевания в соответствии с чем выделяют:
- Периферический нейрофиброматоз (1 тип)— в клинической картине доминируют поражение периферических нервов и кожных покровов.
- Центральный нейрофиброматоз (2 тип) — в патологический процесс вовлекаются преимущественно черепные нервы и спинальные корешки.
Причины
Этиологическим фактором нейрофиброматоза 1 типа являются множественные мутации одного из самых протяженных и сложно организованных генов NF1 (17q11.2), являющихся следствием инактивации преимущественно отцовской аллели. Причиной нейрофиброматоза 2 типа является генетический дефект в 22 хромосоме (22q12).
Симптомы
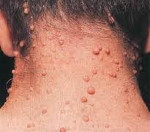
Нейрофиброматоз Реклингхаузена начинает проявляться в большинстве случаев множественными нейрофибромами, с локализацией по ходу периферических нервов, в виде округлых болезненных узелков, расположенных в толще кожи, различающиеся по своим размерам. Нейрофибромы представлены в виде узелков на/в толще кожи нормальной или синюшно-красной окраски и мягко эластической консистенции.
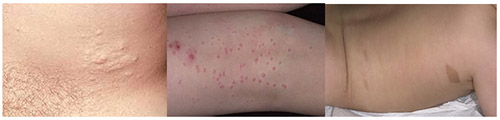
Мягкие кожные опухоли, как правило, у детей отсутствуют, особенно первых лет жизни и возникают к 10-15 годам. При этом, их количество особенно возрастает в пубертатном периоде. При пальпации они безболезненны, однако при вовлечении в патологический процесс периферических нервов, могут возникают боли, гипестезии. На коже могут присутствовать и другие изменения: участки депигментации, сосудистые пятна, очаговое поседение волос, гипертрихоз. Возможно развитие нейрофибром. Плексиформная нейрофиброма может образовывать гигантские опухоли, состоящие из кожных и подкожных элементов.
Некоторые авторы (В.В. Мордовцева) выделяет четыре варианта клинического течения нейрофиброматоза I типа:
Факультативные симптомы нейрофиброматоза проявляются изменениями в костной системе в виде различных деформаций позвоночного столба — сколиозы, лордосколиозы, кифосколиозы, псевдоартрозы, утончение/искривление длинных трубчатых костей, патологических переломов. Нередко развиваются лицевые дисморфии: аномалии глазных щелей, глазной гипертелоризм, деформация ушных раковин, неправильная форма черепа и др. Для пациентов с НФ-I характерны макроцефалия, низкий рост, дисплазия клиновидной кости и затылочной части черепа.
Характерными поражениями нервной системы являются судорожный и гипертензионно-гидроцефальный синдромы, опухоли ЦНС (эпендимома, астроцитома, глиома зрительного нерва/ствола мозга), эпилепсия, что появляется соответствующей симптоматикой. Так, глиома зрительного нерва проявляется нарушением зрения.
Для NF1 характерны дополнительные проявления в виде когнитивных расстройств от легких до выраженных, часто в сочетании с умеренным снижением IQ ребенка, затруднением в освоении чтения, письма, математики и нервно-психической неуравновешенностью; наличие эндокринных расстройств (полового созревания, феохромоцитома, нарушение роста и др.). Со стороны сосудов у пациентов часто регистрируется стеноз почечных артерий с повышением АД, окклюзия артерий, болезнь Мойа-Мойа.
Клиническая манифестация заболевания НФ2 включает развитие двусторонних вестибулярных шванном или шванном других черепных периферических и спинальных нервов при минимальных экстраневральных и кожных симптомах. Двусторонняя шваннома (невринома) слухового нерва относится к основным признакам, который встречается практически в 90% случаев, редко обнаруживается у детей, развиваются преимущественно в позднем подростковом/раннем взрослом возрасте. НФ-II.
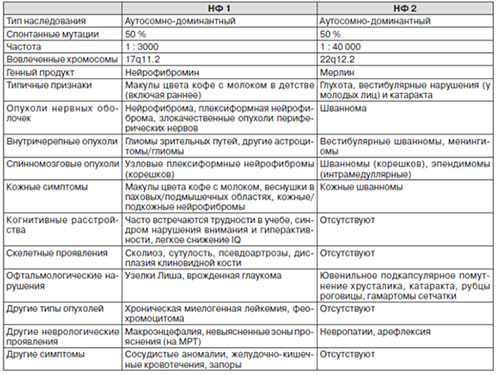
Ниже в таблице приведены характерные проявления нейрофиброматоза 1 и 2 типа.
Анализы и диагностика
Диагноз нейрофиброматоза 1 типа ставится на основе абсолютных и относительных клинических признаков и данных инструментальных методов диагностики:
- КТ/МРТ позвоночника, головного мозга, внутренних органов.
- Рентгенография позвоночника.
- Электрокохлеография.
- Офтальмоскопия.
- Импедансометрия.
- Генетическое обследование и проведение генеалогического анализа.
Лечение
Патогенетическое лечение нейрофиброматоза до настоящего времени не разработано. В большинстве случаев проводится симптоматическая терапия, что и подтверждает специализированный форум. Так, при выраженном болевом синдроме показаны НПВС (Диклофенак, Мелоксикам, Ибупрофен, Нимесулид, Напроксен, Кетопрофен, Пироксикам), неопиодные (Парацетамол, Анальгин, Бутадион, Аспирин и др.) и опиоидные анальгетики (Трамадол, Фентанил, Налбуфин, Тримеперидин), трициклические антидепрессанты (с осторожностью из-за высокого риска судорожного синдрома), Нейронтин (Габапентин), Топирамат. Консервативная терапия предусматривает курсовое назначение препаратов, влияющие на:
- дегрануляцию тканевых базофилов (назначается Кетотифен);
- пролиферацию клеточных элементов (назначаются ретиноиды — Ацитретин, Этретинат, Бексаротен, Тазаротен);
- снижение во внеклеточном матриксе количества гликозаминогликанов (Гиалуронидаза).
Согласно последним рекомендациям, лечение нейрофиброматоза может проводиться с использованием новых перспективных препаратов для таргетной (нацеленной) терапии, воздействующие на определенное звено в патогенетической цепи нейрофиброматоза. Для таргетной терапии используются два вида молекул: мелкие молекулы и моноклональные антитела. К таким препаратам относятся Бевацизумаб (Авастин), Эрлотиниб, Иматиниб, Салиразиб, Лапатиниб (Тайверб).
В случаях малигнизации опухолей проводится химиотерапия и лучевая терапия. При когнитивных нарушениях и плохой обучаемости детей рекомендуется проводить учебный процесс в спецшколах, а также проведение социальной реабилитации взрослых пациентов.
Нейрофиброматозы – наследственные заболевания, характеризующиеся образованием доброкачественных опухолей в коже, мягких тканях, нервной системе и внутренних органах. Выделяют 6 типов нейрофиброматозов, клинически значимы типы I и II. Общие симптомы включают нейрофибромы на коже, опухоли спинномозговых корешков, слуховых и зрительных нервов, пигментные пятна, костные деформации. Диагностика основана на данных осмотра пациентов, выявлении опухолей с помощью МРТ и КТ спинного и головного мозга, внутренних органов. Лечение симптоматическое – проводится резекция опухолей, рентгенотерапия, химиотерапия.
МКБ-10
- Причины нейрофиброматозов
- Патогенез
- Симптомы нейрофиброматозов
- Осложнения
- Диагностика
- Лечение нейрофиброматозов
- Прогноз и профилактика
- Цены на лечение
Общие сведения
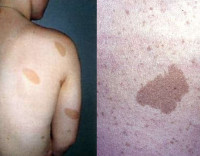
Нейрофибромы – доброкачественные опухоли, развивающиеся из оболочек нервных волокон. Чаще всего располагаются в слоях кожи и подкожной клетчатке, иногда поражают головной мозг, нервные волокна, корешки спинного мозга, мягкие ткани, внутренние органы. Нейрофиброматоз – болезнь, при которой образуются многочисленные нейрофибромы. Распространенность разных типов патологии значительно колеблется: заболеваемость 1 типом составляет 1:2 500, 2 типом – 1:50 000. Другие варианты встречаются еще реже, их точная эпидемиология не определена. Гендерной и расовой предрасположенности не выявлено. Дебют клинических проявлений возможен в любом возрасте, зависит от типа болезни.
Причины нейрофиброматозов
Образование множественных нейрофибром детерминировано генетически. При нейрофиброматозе I существует мутация гена НФ1, расположенного на длинном плече 17 хромосомы. Он относится к генам-супрессорам роста опухолевых тканей, большая часть из которых – нейроэктодермального генеза. При дефекте в гене НФ1 нарушается синтез белков, ответственных за клеточную пролиферацию. Мутации носят характер транслокаций, делеций, инверсий, точковых изменений. Больше 80% из них приводят к синтезу нефункциональных белков или к полному отсутствию белковых молекул. Наследование происходит по аутосомно-доминантному механизму с высокой степенью пенетрации: при наличии мутационного гена у одного из родителей вероятность болезни у ребенка составляет 50%, если оба родителя имеют мутацию, риск повышается до 80-90%. Известны случаи спонтанных мутаций.
Причиной нейрофиброматоза II является мутационное изменение гена НФ2, локализованного на 22 хромосоме. Он кодирует производство белка мерлина (шванномина) – супрессора опухолевого роста. Тип наследования – аутосомно-доминантный с небольшой степенью пенетрации. Передача одного мутантного гена зачастую не проявляется, поскольку второй ген обеспечивает синтез достаточного количества белков. Если он повреждается, синтез нормальных фракций мерлина прекращается, пролиферация клеток усиливается, развивается новообразование. При других типах нейрофиброматозов также существуют мутации в генах, обеспечивающих воспроизведение молекул белков-супрессоров роста опухолей.
Патогенез
Общим патогенетическим механизмом развития нейрофиброматозов является наследственно обусловленная недостаточность какого-либо белка, подавляющего процессы опухолевой пролиферации клеток в тканях нейроэктодермального происхождения. При мутации одного гена производство опухолевых супрессоров прекращается наполовину, равновесие роста и гибели клеток смещается в сторону митотического деления. Нормальный аллельный ген частично компенсирует дефицит белка. Тяжесть нейрофиброматоза определяется тем, насколько дефектный ген влияет на активность белка-супрессора – частично или полностью нарушает функциональность, полностью блокирует производство. Кроме этого, выраженность клинических признаков зависит от сохранности противоопухолевого иммунитета.
Во многих органах и тканях пациентов формируются доброкачественные опухолевые образования, состоящие из соединительной ткани и пигментных клеток. На нервных стволах образуются невриномы; на поверхности кожи – пигментированные области, жировые бляшки, расширенные сосуды; на сетчатке глаз – факоматоз. Изменяется строение костей, они остаются недоразвитыми либо чрезмерно утолщаются, искривляется позвоночный столб.
Симптомы нейрофиброматозов
В период пубертата и позже формируются кожные и плексиформные нейрофибромы, располагающиеся соответственно подкожно (на мелких нервных волокнах, иннервирующих кожу) и на крупных нервах. Они представляют собой небольшие доброкачественные новообразования. Кожные нейрофибромы воспринимаются как косметический дефект, при определенном расположении травмируются. Плексиформные опухоли, локализующиеся по ходу периферических нервов, выявляются на конъюнктиве, веках, в брюшной полости и средостении. Проявляются хронической болью, онемением, судорогами, параличом. Опухоли ЦНС находятся внутри черепа, представлены глиомами зрительных нервных волокон, астроцитомами, эпендимомами, невриномами слухового нерва, менингиомами и нейрофибромами. Клиническая картина определяется размерами новообразований, вовлеченностью мозговых структур в патологический процесс. В детском возрасте диагностируются расстройства психического развития: снижение когнитивных способностей, гиперактивность, редко – деменция.
При тяжелом нейрофиброматозе деформируется костная система. У больных возникает сколиоз, краевые структурные изменения тел позвонков и их отростков, эрозийные поражения краев межпозвоночных отверстий и задних ребер. Характерна атрофия либо, наоборот, гипертрофия трубчатых костей. Кости часто искривлены, на поверхности обнаруживаются периостальные гребни и наслоения. В полостях костей могут образовываться нейрофибромы. Если в процесс вовлекаются кости черепа, появляется внешняя асимметрия, наиболее выраженная при поражении лицевой части и глазниц. Свод черепа может иметь атрофированные участки, дефекты, узуры, иногда отмечается локальное увеличение костного вещества.
При типе 2 формируются высокодифференцированные опухоли, которые, однако, более агрессивны, чем при заболевании 1 типа. Пигментных пятен нет. Образуются невриномы – подвижные и болезненные неоплазии. Нередко они локализуются на слуховом нерве, вызывая потерю слуха. Нейрофиброматоз 3 типа отличается большим количеством нейрофибром, ускоренным развитием нейролемм и глиом зрительного нерва, приводящих к расстройству зрения. Специфический признак – появление нейрофибром на ладонях. При болезни 4 типа симптомы похожи, сохраняется риск поражения зрительных волокон. Для 5 типа характерны пигментные темные пятна, опухоли больших размеров, провоцирующие асимметрию тела. Течение 6 типа сопровождается лишь пигментными пятнами. При 7 типе выявляются нейрофибромы средних размеров, гиперпигментации нет.
Осложнения
В 10% случаев нейрофибромы трансформируются в злокачественные опухоли. В группе высокого риска находятся пациенты с большим катамнестическим стажем, беременные женщины. У 6% детей нарушается умственное развитие: они имеют проблемы при освоении учебных навыков (чтение, письмо, счет), с трудом запоминают новую информацию, долго адаптируются в незнакомых ситуациях. Больные всех возрастов подвержены депрессии, поскольку испытывают дискомфорт, чувство стыда и неловкости из-за обезображенной внешности. Множественные нейрофибромы провоцируют эндокринные расстройства, эпилептические припадки, гипотонию мышц, стеноз почечной и легочной артерии, легочные кисты, интерстициальную пневмонию, гипертрофию клитора, нарушения развития органов ЖКТ.
Диагностика
Подозрение на нейрофиброматоз возникает при множественных подкожных опухолях, пигментных пятнах, спинальной шванноме, ухудшении слуха и зрения. Обследование проводят дерматовенеролог, невролог, офтальмолог, отоларинголог и генетик. Перед инструментальными и лабораторными процедурами осуществляется сбор семейного и личного анамнеза, клинический опрос и осмотр. В ходе генеалогического анализа выявляется передача заболевания в нескольких поколениях, реже определяется первичная спонтанная мутация. На теле пациентов обнаруживаются нейрофибромы, пигментные области (при определенных типах болезни), искривления позвоночника, деформации костей, нарушения зрения, слуха, координации движений. Производится дифференциальная диагностика различных вариантов нейрофиброматозов, исключается синдром Протея, рассеянный липоматоз, синдром Клиппеля-Треноне-Вебера. Для уточнения диагноза назначаются:
- МРТ, КТ. Визуализационные методы исследований позволяют определить наличие нейрофибром в головном мозге, позвоночнике, внутренних органах. Двусторонние невриномы 7 пары черепных нервов являются диагностическим критерием нейрофиброматоза II типа. Часто выявляются глиомы, шванномы, менингиомы. Для I типа свойственно развитие плексиформных и обычных новообразований, глиом.
- Рентгенография костей скелета. Диагностическая процедура выполняется с целью подтверждения и оценки тяжести сколиоза, костных атрофий и гипертрофий, локальных утолщений и эрозийных поражений костных структур. При большинстве типов болезни наблюдается истончение кортикального слоя, ложные суставы, дисплазии крыльев клиновидной кости, дугообразное искривление большеберцовой и малоберцовой костей, кисты длинных костей.
- Офтальмологическое исследование. Нейрофиброматоз типа 1 сопровождается плексиформной нейрофибромой век, меланоцитарными гамартомами радужки, глиомой оптических нервных волокон, астроцитарной гамартомой сетчатки, утолщением роговичных нервов, конъюнктивальной нейрофибромой, ишемическими поражениями венул сетчатки. Патогномоничный признак – пятна светлого оттенка на глазном дне и радужке (гамартомы). При 2 типе диагностируется задняя субкапсулярная катаракта, помутнение хрусталика.
- Аудиологическое исследование. При опухолевом поражении слуховых нервов и жалобах на нарастающую глухоту (тугоухость) выполняется электрокохлеография и импедансометрия. Результаты указывают на снижение остроты слуха, наличие слуховой нейропатии, определяют причину и локализацию нарушения.
Лечение нейрофиброматозов
В настоящее время терапия данной группы заболеваний заключается в симптоматической помощи больным. Пациенты регулярно проходят обследования, нацеленные на контроль формирования и увеличения опухолей. При наличии нейрофибром, провоцирующих боль, расположенных в местах повышенного риска травмирования, сдавливающих или смещающих жизненно важные органы, проводится их хирургическое удаление. Применяются классические методики резекции неоплазий и участков нервов, криодеструкция, лазерная хирургия. При множественных новообразованиях назначается лучевая терапия, химиотерапия. Больным с поражением опорно-двигательного аппарата показаны реабилитационные мероприятия (физиолечение, ЛФК).
Активно разрабатываются способы этиологического лечения нейрофиброматозов. На стадии клинических испытаний находится терапия ингибиторами RAS (белков-активаторов роста опухолей) у лиц с нейрофиброматозом первого типа. Этап теоретических разработок проходят методы генной инженерии. Усилия ученых-генетиков направлены на создание и внедрение в организм больных нормального НФ1 гена, отвечающего за синтез нейрофибромина, на расшифровку и введение гена ФН2, обеспечивающего транскрипцию белка шванномина. В некоторых медицинских центрах предпринимаются попытки применения патогенетической терапии, в основе которой лежит комплексное использование стабилизаторов мембран тучных клеток, антипролиферативных препаратов и ферментов, корректирующих метаболические процессы.
Прогноз и профилактика
Нейрофиброматозы являются прогностически благоприятными заболеваниями – малигнизация опухолей происходит редко, в большинстве случаев больные остаются трудоспособными и социально адаптированными. При правильных и регулярных реабилитационных мероприятиях нарушения со стороны костной системы и задержка умственного развития успешно корректируются. Поскольку заболевание является наследственным, профилактика возможна на этапе планирования беременности, парам из групп риска (с отягощенным семейным анамнезом) рекомендуется медико-генетическое консультирование с определением вероятности рождения больного ребенка.
а) Терминология:
1. Сокращения:
• Нейрофиброматоз 1 типа (НФ1), нейрофиброма корешка спинного мозга (НФ), плексиформная нейрофиброма (ПНФ), злокачественная опухоль оболочек периферического нерва (ЗООПН)
2. Синонимы:
• Болезнь Реклингхаузена, периферический нейрофиброматоз
3. Определения:
• Аутосомно-доминантная мезодермальная дисплазия, характеризующаяся формированием плексиформных нейрофибром и нейрофибром корешков спинного мозга, деформацией позвоночника, неопластическими и неопухолевыми поражениями головного мозга, кожными стигмами
б) Визуализация:
1. Общие характеристики позвоночника при нейрофиброматозе 1 типа:
• Наиболее значимый диагностический признак:
о Кифосколиотическая деформация ± множественные опухоли корешков, плексиформная нейрофиброма, дуральная эктазия/латеральные менингоцеле
• Локализация:
о На всем протяжении краниоспинальной оси
• Размеры:
о Варьируют от мелких до весьма значительных
• Морфология:
о Кифотическая/кифосколиотическая деформация позвоночника зачастую достаточно тяжелая и весьма причудливая по своему виду
о Нейрогенные опухоли, локализованные в корешках спинного мозга, а также в плексиформных объемных образованиях нервов, поражения кожи
3. КТ позвоночника при нейрофиброматозе 1 типа:
• Бесконтрастная КТ:
о Гиподенсное веретеновидное или фокальное увеличение корешков ± гетерогенное объемное образование спинного мозга (глиальная опухоль)
о Дуральная эктазия ± латеральные менингоцеле, плотность которых соответствует СМЖ
• КТ с КУ:
о Опухоли, характеризующиеся легким/умеренным накоплением контраста
• Костная КТ:
о Изменения позвоночника аналогичны таковым, выявленным при рентгенографии, расширение спинномозгового канала и межпозвонковых отверстий на фоне дуральной эктазии ± опухоли спинного мозга
5. Радиоизотопные исследования:
• ПЭТ:
о Стандартный уровень захвата (SUV, standard uptake value) фтордезоксиглюкозы у ЗООПН выше, чем у доброкачественных опухолей
6. Рекомендации по визуализации:
• Наиболее оптимальный метод диагностики:
о МРТ
• Протокол исследования:
о Рентгенография для количественной оценки и определения тактики лечения кифотической, сколиотической деформации позвоночника
о Многоплоскостная МРТ с КУ (особенно в режимах STIR, Т2 FS и T1 с КУ) для оценки состояния спинного мозга, поражения нервов
о Костная КТ для оценки особенностей костной анатомии при предоперационном планировании
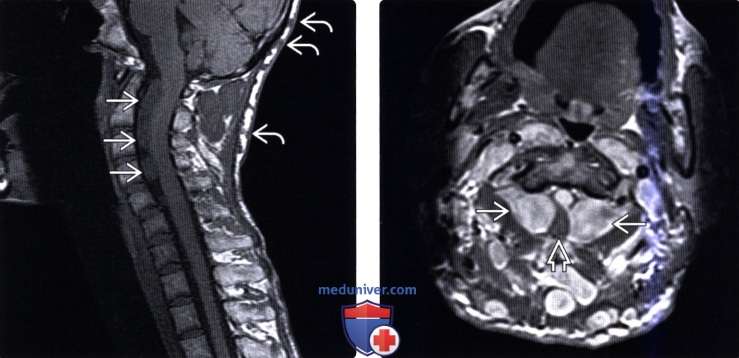
(Слева) Т1-ВИ, сагиттальная проекция: распространенные интрадуральные экстрамедуллярные нейрофибромы, расположенные вдоль вентральной поверхности спинного мозга и оттесняющие его кзади. Множественные подкожные нейрофибромы также видны на задней поверхности шеи (некоторые из них обозначены символом).
(Справа) На аксиальном Т1 с КУ на верхне-шейном уровне опрделяются признаки множественного нейрофиброматозного поражения мягких тканей и позвоночника. Обратите внимание на тяжелую компрессию спинного мозга на уровне С2 двусторонними нейрофибромами.
в) Дифференциальная диагностика поражения позвоночника при нейрофиброматозе 1 типа:
1. Нейрофиброматоз 2 типа (центральный нейрофиброматоз):
• Множественные интракраниальные шванномы и менингиомы, шванномы и менингиомы позвоночника
• Деформации позвоночника наблюдаются нечасто
• Клинические, лабораторные и генетические признаки, характерные для НФ2
3. Врожденные гипертрофические полирадикулонейропатии:
• Болезни Шарко-Мари-Тута, Дежерин-Сотта
• Увеличение размеров корешков спинного мозга напоминает ПНФ
• Характерные для НФ1 кожные стигмы отсутствуют
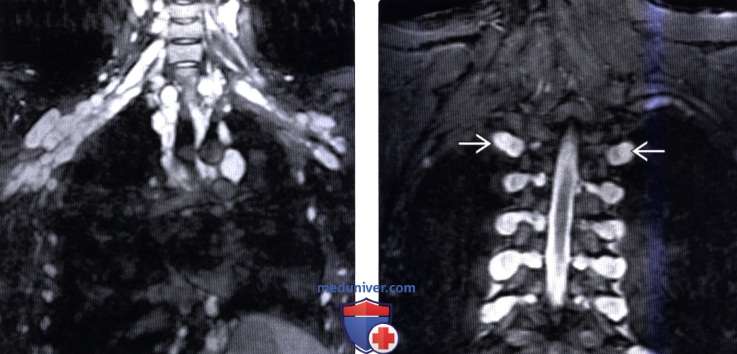
(Слева) На фронтальном STIR МР-И визуализируются множественные плексиформные нейрофибромы, поражающие с двух сторон спинномозговые нервы, симпатические стволы и плечевые сплетения, а также межреберные нервы.
(Справа) На фронтальном STIR МР-И определяются признаки двустороннего нейрофиброматозного поражения грудных корешков спинного мозга, опухоли распространяются через межпозвонковые отверстия в паравертебральные мягкие ткани. В режиме STIR нейрофибромы отличаются умеренной гиперинтенсивностью сигнала.
г) Патология:
4. Микроскопия:
• Опухоли образованы располагающимися вдоль нервных волокон шванновскими клетками + периневральными фибробластами
о Коллагеновые волокна, мукоидный/миксоидный матрикс, опухолевые клетки, пучки нервных волокон
о Положительный результат иммуногистохимической окраски на S100 протеин, фигуры митоза в отсутствие малигнизации опухоли наблюдаются редко
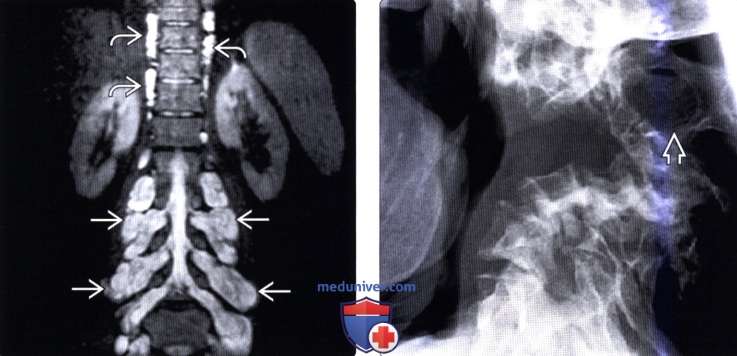
(Слева) На аксиальном STIR МР-И визуализируются множественные двусторонние нейрофибромы поясничных корешков спинного мозга, распространяющиеся через межпозвонковые отверстия в окружающие паравертебральные мягкие ткани. Также здесь обнаружены небольшие плексиформные нейрофибромы симпатических стволов.
(Справа) Рентгенограмма шейного отдела позвоночника в боковой проекции: необычная и тяжелая остроугольная кифотическая деформация шейного отдела и увеличение размеров межпозвонкового отверстия - очень характерные для НФ1 изменения.
д) Клинические особенности изменений позвоночника при нейрофиброматозе 1 типа:
2. Демография:
• Возраст:
о Диагноз обычно ставится в детстве, однако при минимальной выраженности изменений диагноз может быть выставлен уже во взрослом возрасте
• Пол:
о М = Ж
• Этническая принадлежность:
о Увеличение частоты в арабо-израильских популяциях
• Эпидемиология:
о Часто (1:4000)
3. Течение заболевания и прогноз:
• Кифотическая, сколиотическая деформация позвоночника часто прогрессирует
• Сами НФ обычно растут медленно, ускорение их роста отмечается при беременности, в период полового созревания или при малигнизации
4. Лечение позвоночника при нейрофиброматозе 1 типа:
• Консервативное наблюдение, необходимость вмешательства диктуется имеющейся клинической симптоматикой и характером новообразований
• Хирургическая резекция клинически значимых локализованных ИФ, опухолей спинного мозга
• Инвазивные ПНФ редко являются резектабельными; наблюдение ± био- или химиотерапия (талидомид, антигистаминные препараты, факторы созревания, антиангиогенные препараты)
• Спондилодез применяется только при клинически значимых или тяжелых деформациях позвоночника
е) Диагностическая памятка:
1. Следует учесть:
• Множественные опухоли оболочек нервов, > одной нейрофибромы, причудливого вида кифосколиоз с деформацией позвонков → думайте о НФ1
• Отсутствие видимых кожных стигм не исключает НФ1
2. Советы по интерпретации изображений:
• Характерные лучевые признаки плексиформного НФ лучше всего видны в режимах Т2 FS и STIR
ж) Список использованной литературы:
1. Nguyen R et al: Characterization of spinal findings in children and adults with neurofibromatosis type 1 enrolled in a natural history study using magnetic resonance imaging. J Neurooncol. ePub, 2014
2. Pourtsidis A et al: Malignant peripheral nerve sheath tumors in children with neurofibromatosis type 1. Case Rep Oncol Med. 2014:843749, 2014
3. Jett К et al: Clinical and genetic aspects of neurofibromatosis 1. Genet Med. 12(1)4-11,2010
4. Wasa J et al: MRI features in the differentiation of malignant peripheral nerve sheath tumors and neurofibromas. AJR Am J Roentgenol. 194(6): 1 568-74, 2010
5. Scalzone M et al: Neurofibromatosis type 1 clinical features and management. Pediatr Med Chir. 31 (6):246-51, 2009
6. Van Meerbeeck SF et al: Whole body MR imaging in neurofibromatosis type 1. EurJ Radiol. 69(2):236-42, 2009
Редактор: Искандер Милевски. Дата публикации: 23.7.2019
Читайте также:
